Andermann-syndroom: Afwijkingen aan spieren en hersenen
 Bij het Andermann-syndroom komen hersenafwijkingen in combinatie met spier- en gewrichtsproblemen voor. Deze erfelijke ziekte brengt progressieve schade toe aan het zenuwstelsel. De symptomen van het Andermann-syndroom verschijnen reeds vroeg in het leven. De behandeling verloopt moeilijk, en door de progressieve aard van de ziekte komen de meeste patiënten vroegtijdig te overlijden. Het syndroom kent voorts een hoge prevalentie in de Frans-Canadese bevolking in Québec. Naiman en Fraser beschreven het syndroom voor het eerst in 1955. In 1972 beschreven Andermann et al. de aandoening meer in detail.
Bij het Andermann-syndroom komen hersenafwijkingen in combinatie met spier- en gewrichtsproblemen voor. Deze erfelijke ziekte brengt progressieve schade toe aan het zenuwstelsel. De symptomen van het Andermann-syndroom verschijnen reeds vroeg in het leven. De behandeling verloopt moeilijk, en door de progressieve aard van de ziekte komen de meeste patiënten vroegtijdig te overlijden. Het syndroom kent voorts een hoge prevalentie in de Frans-Canadese bevolking in Québec. Naiman en Fraser beschreven het syndroom voor het eerst in 1955. In 1972 beschreven Andermann et al. de aandoening meer in detail.
- Synoniemen Andermann-syndroom
- Epidemiologie syndroom van Andermann
- Oorzaken en erfelijkheid
- Symptomen: Afwijkingen aan spieren en gewrichten, hersenen en gezicht
- Diagnose en onderzoeken
- Behandeling aandoening
- Prognose: Progressieve ziekte
Synoniemen Andermann-syndroom
Het Andermann-syndroom (ACCPN, AS, HMSN / ACC) is eveneens gekend onder deze synoniemen:- agenesie van corpus callosum met neuronopathie
- agenesie van corpus callosum met perifere neuropathie
- agenesie van corpus callosum met polyneuropathie (verminderd pijngevoel, verlamming, prikkelingen en tintelingen in handen en voeten)
- erfelijke motorische en sensorische neuropathie met agenesie van het corpus callosum
- ziekte van Charlevoix
Epidemiologie syndroom van Andermann
Het Andermann-syndroom komt het vaakst voor in de Frans-Canadese bevolking van de Saguenay-Lac-St.-Jean en de Charlevoix regio van noordoostelijk Quebec. Hierbij presenteert het Andermann-syndroom zich bij bijna 1 op de 2000 pasgeborenen. Veel getroffen patiënten zijn afkomstig van een gemeenschappelijk voorouderlijk koppel uit Frankrijk, dat in 1657 in Québec City getrouwd is. Slechts enkele patiënten met deze aandoening zijn geïdentificeerd in andere delen van de wereld.Oorzaken en erfelijkheid
Genmutatie
Mutaties (= veranderingen) in het SLC12A6-gen veroorzaken het Andermann-syndroom.Overervingspatroon
Deze aandoening kent een autosomaal recessief overervingspatroon, waardoor beide kopieën van het gemuteerde gen in elke cel aanwezig moeten zijn opdat de aandoening tot uiting komt. De ouders van een patiënt met een autosomaal recessieve aandoening dragen elk een exemplaar van het gemuteerde gen, maar hebben vrijwel nooit symptomen van het syndroom.Symptomen: Afwijkingen aan spieren en gewrichten, hersenen en gezicht
Het Andermann-syndroom is een aandoening die motorische en sensorische neuropathie met zich meebrengt. Bij de patiënt is met andere woorden zenuwen beschadigd die nodig zijn voor spierbewegingen en gevoel. Afwezigheid (agenesie) of misvorming van het weefsel dat de linker- en rechterhelft van de hersenen met elkaar verbindt, komt ook voor. Bij deze aandoening geldt dat de spier- en gewrichtsproblemen en hersenafwijkingen vaak tot uiting komen, maar de ernst, uitgebreidheid en aanwezigheid van de andere symptomen is sterk verschillend bij patiënten, zelfs onder dezelfde familie.Spieren en gewrichten
Patiënten die getroffen zijn door het Andermann-syndroom hebben abnormale of afwezige reflexen (areflexie) en een zwakke spierspanning (hypotonie). Zij ervaren spieratrofie (spierverlies), een ernstige progressieve zwakte en een verlies van gevoel in de ledematen, en ritmische trillingen (tremor: bevingen). De patiënten beginnen meestal met lopen wanneer ze tussen de drie en vier jaar zijn maar door de progressieve aard van de aandoening, verliezen ze dit vermogen in de tienerjaren. Op latere leeftijd ontwikkelen patiënten contracturen (aanhoudende spierspanning), waardoor de beweging van bepaalde gewrichten beperkt is. Daarnaast vertonen de meeste patiënten zich met een abnormale kromming van de wervelkolom (scoliose), waarvoor een operatie nodig is. De grote teen kruist bovendien soms over de andere tenen. Daarnaast zijn de tweede en derde teen gedeeltelijk gefuseerd (syndactylie).Hersenen
Het Andermann-syndroom is eveneens gekenmerkt door hersenafwijkingen. Het corpus callosum ontbreekt namelijk bij de meeste patiënten. Dit is een structuur die de rechter- en linkerzijde van de hersenen met elkaar verbindt. Dit leidt tot spierzwakte in het gezicht, hangende oogleden (ptosis), en oftalmoplegie (spierverlamming in de ogen). Patiënten met het Andermann-syndroom hebben meestal een milde tot ernstige verstandelijke handicap en ook epileptische aanvallen komen voor. Daarnaast ontwikkelt de patiënt vaak psychiatrische symptomen in het tweede decennium, zoals depressie, angst, agitatie (opwinding), paranoia en hallucinaties, die gewoonlijk verschijnen in de adolescentie. De patiënt heeft voorts mogelijk microcefalie (een erg klein hoofd) en soms ook craniosynostose.Gezicht
Sommige patiënten met het Andermann-syndroom hebben atypische fysieke kenmerken, zoals ver uit elkaar geplaatste ogen (hypertelorisme), een brede, korte schedel (brachycefalie), een klein hoofd (microcefalie), een lang asymmetrisch gezicht, een kleine bovenkaak en grote oren.Ogen
Ooggerelateerde symptomen omvatten myopie (bijziendheid), nystagmus (wiebelogen) en strabisme (scheelzien).Diagnose en onderzoeken
Lichamelijk onderzoek
De patiënt vertoont typische afwijkingen die de arts noteert. Deze wijzen reeds in de richting van het syndroom.Diagnostisch onderzoek
Een elektro-encefalografie (EEG: hersenfilmpje) laat afwijkingen zien. Een genetisch onderzoek bevestigt de diagnose.Behandeling aandoening
Voor het Andermann-syndroom is geen genezing mogelijk. Ook zijn weinig effectieve behandelingen beschikbaar om de symptomen te verlichten. Fysiotherapie in combinatie met chirurgie om de wervelkolom recht te zetten, zijn de enige behandelingsmogelijkheden.Prognose: Progressieve ziekte
Het Andermann-syndroom is een progressieve ziekte waarbij de motorische functies van een patiënt zijn aangetast. Alle patiênten hebben een verstandelijke handicap die varieert in ernst, en ook iedereen belandt uiteindelijk in een rolstoel. Ernstige psychische problemen ontstaan in het tweede decennium. De meeste patiënten met het Andermann-syndroom hebben een kortere levensduur. Veelal halen ze echter wel de volwassenheid en overlijden ze rond het vierde decennium.© 2016 - 2024 Miske, het auteursrecht van dit artikel ligt bij de infoteur. Zonder toestemming is vermenigvuldiging verboden. Deze informatie is van informatieve aard en geen vervanging voor professioneel medisch advies. Per 2021 gaat InfoNu verder als archief, artikelen worden nog maar beperkt geactualiseerd.
 Alpers Huttenlocher-syndroom: Neurodegeneratieve aandoeningHet Alpers Huttenlocher-syndroom is een neurodegeneratieve aandoening waarbij de hersenen en motoriek aftakelen. Daarnaa…
Alpers Huttenlocher-syndroom: Neurodegeneratieve aandoeningHet Alpers Huttenlocher-syndroom is een neurodegeneratieve aandoening waarbij de hersenen en motoriek aftakelen. Daarnaa…
 Perry-syndroom: Progressieve neurologische aandoeningHet uiterst zeldzame Perry-syndroom is een progressieve neurologische aandoening waarbij een patiënt kampt met bewegings…
Perry-syndroom: Progressieve neurologische aandoeningHet uiterst zeldzame Perry-syndroom is een progressieve neurologische aandoening waarbij een patiënt kampt met bewegings…
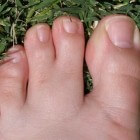 Syndactylie: Samengegroeide vingers en/of tenenSyndactylie verwijst naar een aangeboren fusie van twee of meer vingers en/of tenen. Normaal gezien scheiden de vingers…
Syndactylie: Samengegroeide vingers en/of tenenSyndactylie verwijst naar een aangeboren fusie van twee of meer vingers en/of tenen. Normaal gezien scheiden de vingers…
 Mastocytose: Huidaandoening met ophoping van mestcellenMastocytose is een zeldzame aandoening waarbij abnormale accumulaties (ophopingen) van mestcellen in de huid, het beenme…
Mastocytose: Huidaandoening met ophoping van mestcellenMastocytose is een zeldzame aandoening waarbij abnormale accumulaties (ophopingen) van mestcellen in de huid, het beenme…
 Allan-Herndon-Dudley syndroom: Neuromusculaire aandoeningHet Allan-Herndon-Dudley syndroom is een neuromusculaire aandoening waarbij de patiënt een verstandelijke beperking en b…
Allan-Herndon-Dudley syndroom: Neuromusculaire aandoeningHet Allan-Herndon-Dudley syndroom is een neuromusculaire aandoening waarbij de patiënt een verstandelijke beperking en b…
Gerelateerde artikelen
Interessante informatie over het syndroom van DownHet syndroom van Down komt veel voor in Nederland. Maar wat is het eigenlijk precies voor soort aandoening?
Bronnen en referenties
- AGENESIS OF THE CORPUS CALLOSUM WITH PERIPHERAL NEUROPATHY; ACCPN, http://www.omim.org/entry/218000, geraadpleegd op 23 juni 2016
- Andermann Syndrome, http://www.medlink.com/article/andermann_syndrome, geraadpleegfd op 23 juni 2016
- Andermann syndrome, https://ghr.nlm.nih.gov/condition/andermann-syndrome#synonyms, geraadpleegd op 23 juni 2016
- Andermann Syndrome, https://www.counsyl.com/services/family-prep-screen/diseases/andermann-syndrome/, geraadpleegd op 23 juni 2016
- Overview, https://rarediseases.info.nih.gov/gard/1537/andermann-syndrome/resources/1, geraadpleegd op 23 juni 2016
- Symptoms, https://rarediseases.info.nih.gov/gard/1537/andermann-syndrome/resources/9, geraadpleegd op 23 juni 2016
Miske (4.039 artikelen)
Laatste update: 06-04-2020
Rubriek: Mens en Gezondheid
Subrubriek: Aandoeningen
Bronnen en referenties: 6
Laatste update: 06-04-2020
Rubriek: Mens en Gezondheid
Subrubriek: Aandoeningen
Bronnen en referenties: 6
Per 2021 gaat InfoNu verder als archief. Het grote aanbod van artikelen blijft beschikbaar maar er worden geen nieuwe artikelen meer gepubliceerd en nog maar beperkt geactualiseerd, daardoor kunnen artikelen op bepaalde punten verouderd zijn. Reacties plaatsen bij artikelen is niet meer mogelijk.
Medische informatie…
Deze informatie is van informatieve aard en geen vervanging voor professioneel medisch advies. Raadpleeg bij medische problemen en/of vragen altijd een arts.
Deze informatie is van informatieve aard en geen vervanging voor professioneel medisch advies. Raadpleeg bij medische problemen en/of vragen altijd een arts.