Wiskott-Aldrich-syndroom: Problemen met bloedstolling & huid
 Het Wiskott-Aldrich-syndroom is een aangeboren aandoening waarbij voornamelijk mannelijke patiënten problemen krijgen met het immuunsysteem en het bloed. Hierdoor komen infecties en bloedingen vaker voor. De ernstige ziekte vereist vaak een agressieve behandeling zoals een beenmergtransplantatie. Andere ondersteunende behandelingen zijn ook mogelijk. Ondanks verschillende therapieën blijft de levensverwachting voor patiënten met deze erfelijke ziekte verkort. Het syndroom werd voor het eerst in de medische literatuur vermeld in 1937 door Alfred Wiskott, een Duitse kinderarts. In 1954 werd het syndroom verder in detail beschreven door Robert Aldrich, een Amerikaanse kinderarts.
Het Wiskott-Aldrich-syndroom is een aangeboren aandoening waarbij voornamelijk mannelijke patiënten problemen krijgen met het immuunsysteem en het bloed. Hierdoor komen infecties en bloedingen vaker voor. De ernstige ziekte vereist vaak een agressieve behandeling zoals een beenmergtransplantatie. Andere ondersteunende behandelingen zijn ook mogelijk. Ondanks verschillende therapieën blijft de levensverwachting voor patiënten met deze erfelijke ziekte verkort. Het syndroom werd voor het eerst in de medische literatuur vermeld in 1937 door Alfred Wiskott, een Duitse kinderarts. In 1954 werd het syndroom verder in detail beschreven door Robert Aldrich, een Amerikaanse kinderarts.- Synoniemen Wiskott-Aldrich-syndroom
- Epidemiologie van de aandoening
- Mechanisme
- Oorzaken van de ziekte
- Genetische mutatie
- Overerving
- Risicofactoren
- Risicogroepen
- Symptomen: Problemen met bloedstolling en immuunsysteem
- Bloed
- Immuunsysteem
- Alarmsymptomen
- Diagnose en onderzoeken
- Diagnostisch onderzoek
- Differentiële diagnose
- Behandeling via transplantatie
- Agressieve behandeling
- Ondersteunende behandeling
- Prognose en complicaties
- Complicaties
- Preventie
- Praktische tips voor het leven met / omgaan met wiskott-aldrich-syndroom
- Zorg voor regelmatige medische controles
- Voorkom infecties door goede hygiëne
- Zorg voor een gezonde voeding voor een sterker immuunsysteem
- Bescherm jezelf tegen bloedingen
- Emotionele steun en psychologische zorg
- Zorg voor bescherming tegen ziekteverwekkers
- Misvattingen rond wiskott-aldrich-syndroom
- Wiskott-Aldrich-syndroom komt alleen voor bij mannen
- Het Wiskott-Aldrich-syndroom veroorzaakt alleen huidproblemen, zoals huidaandoeningen en uitslag
- Wiskott-Aldrich-syndroom is te behandelen met eenvoudige medicatie zoals pijnstillers
- Wiskott-Aldrich-syndroom is een allergie die verward kan worden met andere huidaandoeningen
- Wiskott-Aldrich-syndroom kan genezen door een verandering in het dieet of gezond voedingspatroon
- Kinderen met het Wiskott-Aldrich-syndroom zullen altijd een kortere levensverwachting hebben
- Wiskott-Aldrich-syndroom heeft geen invloed op de mentale gezondheid van patiënten
Synoniemen Wiskott-Aldrich-syndroom
De volgende synoniemen zijn bekend voor het Wiskott-Aldrich-syndroom:- eczeem-trombocytopenie-immunodeficiëntie-syndroom
- immunodeficiëntie 2
- Wiskott-syndroom
“IMD 2” is een afkorting die in de medische literatuur wordt gebruikt voor de ziekte.
Epidemiologie van de aandoening
Prevalentie en incidentieHet Wiskott-Aldrich-syndroom (WAS) is een zeldzame erfelijke aandoening die meestal mannen treft, omdat het wordt overgedragen via een X-gebonden recessief patroon. De prevalentie wordt geschat op ongeveer 1 op 1.000.000 mannen wereldwijd, hoewel de daadwerkelijke incidentie moeilijk te bepalen is, aangezien het syndroom vaak niet meteen wordt gediagnosticeerd.
Leeftijd van verschijnselen
De symptomen van het Wiskott-Aldrich-syndroom verschijnen meestal in de vroege kinderjaren, vaak tussen de 6 maanden en 2 jaar. Veel kinderen ontwikkelen aanvankelijk herhaalde infecties door immuundeficiëntie en ook hematologische problemen, zoals trombocytopenie (een laag aantal bloedplaatjes).
Mechanisme
Genetische oorzaakHet Wiskott-Aldrich-syndroom wordt veroorzaakt door mutaties in het WAS-gen, dat codeert voor het eiwit WASp. Dit eiwit speelt een cruciale rol in de regulatie van het cytoskelet in de cellen van het immuunsysteem, zoals T-cellen, B-cellen en bloedplaatjes. Een defect in dit eiwit leidt tot de typische symptomen van de aandoening, waaronder een verzwakt immuunsysteem, bloedingsproblemen en eczeem.
Cellulaire processen
Het defect in WASp leidt tot een verminderde functie van de T-cellen en B-cellen, die essentieel zijn voor de immuunrespons. Hierdoor zijn patiënten vatbaarder voor infecties, vooral bacteriële en virale infecties. Ook is de aanmaak van bloedplaatjes in het beenmerg verstoord, wat resulteert in trombocytopenie en verhoogd risico op bloedingen.
Oorzaken van de ziekte
Genetische mutatie
Het Wiskott-Aldrich-syndroom is het gevolg van een abnormaal werkend WASP-eiwit. Een genetische mutatie in het WAS-gen is verantwoordelijk voor deze gestoorde eiwitfunctie. Door de genmutatie werken de bloedplaatjes niet goed en zijn ze abnormaal klein.Overerving
De ziekte kent een X-gebonden overervingspatroon waarbij het foute gen op het X-chromosoom ligt. Mannen hebben slechts één X-chromosoom en daarom leidt een mutatie op dit chromosoom altijd tot de ziekte.Risicofactoren
Erfelijke factorenDe belangrijkste risicofactor voor het Wiskott-Aldrich-syndroom is het X-gebonden recessieve erfelijkheidsmechanisme. Dit betekent dat de aandoening bijna uitsluitend voorkomt bij mannen, terwijl vrouwen meestal drager zijn zonder symptomen te vertonen. Een mannelijke patiënt heeft een 50% kans om het syndroom te erven van een vrouwelijke drager.
Genetische variaties
Hoewel het meeste geval van Wiskott-Aldrich-syndroom wordt veroorzaakt door mutaties in het WAS-gen, kunnen er ook verschillende genetische variaties zijn die de ernst van de symptomen beïnvloeden. Deze variaties kunnen bepalen hoe goed het WASp-eiwit functioneert, wat invloed heeft op de mate van immunodeficiëntie en andere symptomen.
Risicogroepen
Mannen met X-gebonden erfelijkheidMannen die een vrouwelijke drager als moeder hebben, lopen een risico van 50% om het Wiskott-Aldrich-syndroom te ontwikkelen. Vrouwen die drager zijn van de genetische mutatie hebben een lager risico op het ontwikkelen van symptomen, maar ze kunnen de aandoening aan hun zonen doorgeven.
Patiënten met een familiegeschiedenis van WAS
Gezinnen met een geschiedenis van het Wiskott-Aldrich-syndroom, vooral wanneer meerdere familieleden zijn getroffen, moeten extra aandacht besteden aan de vroege symptomen bij pasgeborenen en kinderen. Genetisch advies kan nuttig zijn voor dragers om te begrijpen wat de risico's zijn voor toekomstige kinderen.
Symptomen: Problemen met bloedstolling en immuunsysteem
Bij deze aandoening komen vooral bij jongens problemen met het bloed en het immuunsysteem voor. Meisjes lijden soms ook aan de ziekte, maar zij ervaren doorgaans mildere symptomen. De ziekte is aangeboren en de eerste tekenen van de ziekte manifesteren zich kort na de geboorte, maar soms ook enkele weken tot maanden later.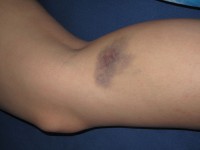 Blauwe plekken komen sneller voor bij het syndroom /
Blauwe plekken komen sneller voor bij het syndroom / Bloed
Bloedingsgerelateerde symptomen treden op omdat patiënten met deze ziekte lijden aan microthrombocytopenie. Dit is de medische term voor een afname van het aantal en de grootte van bloedplaatjes die nodig zijn bij de stolling. Veelvoorkomende bloedstoornissen bij een mannelijke baby of peuter zijn onder andere:- blauwe plekken
- bloederige diarree
- bloedingen in de schedel (door de bevalling)
- bloedingen na een besnijdenis
- bloedingen uit de navelstreng
- kleine puntbloedingen (petechiën) op de huid
- langdurige bloedingen na een relatief klein trauma
- neusbloedingen
- tandvleesbloedingen
- andere bloedingen
Immuunsysteem
Het immuunsysteem werkt bij het Wiskott-Aldrich-syndroom niet goed door een verlaagd aantal witte bloedcellen. Hierdoor ontstaan sneller diverse immuun- en ontstekingsziekten. In de vroege kindertijd ontwikkelt de patiënt matige tot ernstige atopische dermatitis (atopisch eczeem: huidziekte met jeuk). Bij deze chronische huidziekte ontstaan rode, geïrriteerde, droge en jeukende plekken op de huid. Door het verzwakte immuunsysteem komen ook sneller terugkerende infecties (virussen, bacteriën en schimmels) voor, vooral in de oren (otitis media: middenoorontsteking), de sinussen (sinusitis) en de longen (pneumonie: longontsteking).Alarmsymptomen
Herhaalde infectiesEen van de eerste tekenen van het Wiskott-Aldrich-syndroom is de aanleg voor herhaalde infecties, vooral bacteriële en virale infecties. Dit komt door de immuundeficiëntie die de aandoening veroorzaakt. Infecties kunnen ernstig zijn, vooral als ze niet snel worden behandeld.
Bloedingsproblemen en blauwe plekken
Patiënten met WAS vertonen vaak gemakkelijk blauwe plekken en kunnen last hebben van bloedingen, zoals neusbloedingen of bloedingen uit het tandvlees. Dit komt door de trombocytopenie, of het lage aantal bloedplaatjes, die de bloedstolling bemoeilijkt.
Huiduitslag en eczeem
Eczeem is een veelvoorkomend symptoom van het Wiskott-Aldrich-syndroom. Dit kan zich manifesteren als jeukende, droge huid met ontstekingen, vooral op de handen en voeten. Het kan moeilijk te behandelen zijn vanwege de onderliggende immuundeficiëntie.
 Een bloedonderzoek is nodig bij het Wiskott-Aldrich-syndroom / Bron: Frolicsomepl, Pixabay
Een bloedonderzoek is nodig bij het Wiskott-Aldrich-syndroom / Bron: Frolicsomepl, PixabayDiagnose en onderzoeken
Diagnostisch onderzoek
Een huidbiopsie is vereist bij patiënten met het Wiskott-Aldrich-syndroom. Daarnaast is een bloedonderzoek nodig omdat de weefsels histologisch niet te onderscheiden zijn van atopische dermatitis. Hierbij bemerkt de arts het verlaagde aantal bloedplaatjes, die tevens kleiner zijn dan normaal.Differentiële diagnose
De symptomen van het Wiskott-Aldrich-syndroom lijken soms op het klinisch beeld van andere ziekten, zoals:- atopisch eczeem
- Bruton agammaglobulinemie
- het Di George-syndroom
- histiocytose (Langerhans celhistiocytose)
- hypogammaglobulinemie
- idiopathische trombocytopenische purpura (ITP)
- immunodeficiëntie
- neonatale allo trombocytopenie
- trombotische trombocytopenische purpura (TTP)
- X-gebonden ernstige gecombineerde immunodeficiëntie
- X-gebonden lymfoproliferatief syndroom
De definitieve diagnose van de aangeboren aandoening gebeurt aan de hand van genetisch onderzoek.
Behandeling via transplantatie
Agressieve behandeling
Een snelle en agressieve behandeling is nodig om de overlevingskansen te verhogen. De arts kan besluiten om de milt te verwijderen (splenectomie), wat de hoeveelheid bloedplaatjes verhoogt. In sommige gevallen is een beenmergtransplantatie curatief. Een succesvolle beenmergtransplantatie verhoogt in veel gevallen de overlevingskansen aanzienlijk. Onbehandeld overleven deze patiënten de kindertijd meestal niet.Ondersteunende behandeling
Ondersteunende behandelingen zijn nodig om zowel de bloed- als de immuunstoornissen onder controle te houden.Bloed
Bij ernstige trombocytopenie is intraveneus (via een ader) immunoglobuline vereist. Voor ernstige bloedingsstoornissen kan een bloedtransfusie nodig zijn. Verder moeten patiënten voorzorgsmaatregelen nemen om letsels te voorkomen die tot bloedingen kunnen leiden. Kinderen met een zeer laag aantal bloedplaatjes dragen bijvoorbeeld het best een helm om een bloeding in de hersenen te voorkomen.
 Diverse medicijnen bestrijden infecties / Bron: Stevepb, Pixabay
Diverse medicijnen bestrijden infecties / Bron: Stevepb, PixabayCorticosteroïden (krachtige ontstekingsremmers), verzachtende middelen en goede huidverzorging zijn nodig bij eczeem. Sommige patiënten krijgen soms preventief antibiotica om infecties te voorkomen, maar deze medicijnen zijn uiteraard ook inzetbaar bij een infectie zelf.
Prognose en complicaties
Bijna driekwart van de getroffen patiënten hebben een auto-immuun- of ontstekingscomplicatie, waarbij hemolytische anemie (voortijdige afbraak van rode bloedcellen) het vaakst optreedt. Patiënten hebben ook terugkerende infecties, wat de levenskwaliteit vermindert. Het syndroom is progressief en kent een verminderde levensverwachting. Zonder behandeling komt een patiënt meestal te overlijden in het tweede of derde decennium van het leven als gevolg van infecties zoals sepsis (bloedvergiftiging) en meningitis (ontsteking van de hersen- en ruggenmergvliezen), bloedingen of kanker. Leukemie en een B-cel lymfoom zijn de belangrijkste kankersoorten die bij ongeveer 10% van de patiënten voorkomen. Dankzij verfijnde transplantatietechnieken en vroegtijdige splenectomie zijn de overlevingskansen verbeterd, waardoor veel patiënten tegenwoordig een betere levensverwachting hebben.Complicaties
Infecties en sepsisVanwege de verzwakte immuunfunctie kunnen patiënten met Wiskott-Aldrich-syndroom vatbaar zijn voor ernstige infecties, waaronder sepsis. Dit kan levensbedreigend zijn als het niet tijdig wordt herkend en behandeld. De infecties kunnen zowel bacterieel als viraal van aard zijn.
Auto-immuunziekten
Patiënten met WAS kunnen een verhoogd risico hebben op het ontwikkelen van auto-immuunziekten, zoals lupus of artritis. Dit komt doordat het verzwakte immuunsysteem soms een fout maakt en gezonde cellen aanvalt.
Bloedingsstoornissen
De trombocytopenie kan leiden tot ernstige bloedingsproblemen, waaronder inwendige bloedingen en orgaanbeschadiging. Behandeling vereist vaak het beheersen van bloedingen en het corrigeren van het bloedplaatjesaantal, soms door middel van bloedtransfusies of medicijnen.
Preventie
Vroegtijdige diagnose en genetisch adviesAangezien Wiskott-Aldrich-syndroom een erfelijke aandoening is, kunnen genetische testen worden uitgevoerd bij familieleden van een patiënt om de aandoening vroegtijdig op te sporen. Vroegtijdige diagnose kan helpen bij het beheren van symptomen en het voorkomen van complicaties. Genetisch advies is belangrijk voor gezinnen die risico lopen op het doorgeven van de aandoening.
Vaccinaties en infectiebeheersing
Omdat patiënten met WAS een verhoogd risico lopen op infecties, moeten ze zorgvuldig worden gecontroleerd en moeten vaccinaties, zoals voor pneumokokken en griep, op tijd worden gegeven om infecties te voorkomen. Het vermijden van blootstelling aan besmettelijke ziekten is ook van cruciaal belang.