Chediak-Higashi-syndroom: Albinisme met leukocytendefect
 Het Chediak-Higashi-syndroom is een zeldzame multisystemische genetische aandoening die wordt gekenmerkt door een verminderde werking van het immuunsysteem, hypopigmentatie van de huid, ogen en het haar, en bloedstollingsproblemen. Het syndroom werd voor het eerst beschreven door de Cubaanse arts en serologist Alejandro Moisés Chediak en de Japanse kinderarts Otokata Higashi.
Het Chediak-Higashi-syndroom is een zeldzame multisystemische genetische aandoening die wordt gekenmerkt door een verminderde werking van het immuunsysteem, hypopigmentatie van de huid, ogen en het haar, en bloedstollingsproblemen. Het syndroom werd voor het eerst beschreven door de Cubaanse arts en serologist Alejandro Moisés Chediak en de Japanse kinderarts Otokata Higashi.- Synoniemen Chediak-Higashi-syndroom
- Epidemiologie aandoening
- Zeldzaamheid en genetische oorzaken
- Geografische en etnische verschillen
- Mechanisme
- Leukocytendefect en immuundeficiëntie
- Albinisme en visuele afwijkingen
- Oorzaken en erfelijkheid
- Risicofactoren
- Erfelijke aanleg en familiegeschiedenis
- Etnische prevalentie
- Risicogroepen
- Familieleden van dragers
- Mensen van specifieke etnische afkomst
- Symptomen: Albinisme met leukocytendefect
- Alarmsymptomen
- Herhaalde infecties
- Neurologische en hematologische complicaties
- Diagnose en onderzoeken
- Behandeling
- Prognose van de aandoening
- Complicaties
- Preventie
- Praktische tips voor het leven met Chediak-Higashi-syndroom
- Regelmatige controleafspraken en medische opvolging
- Bescherming tegen infecties
- Oogzorg en bescherming tegen licht
- Evenwichtig voedingspatroon en vitamine-inname
- Fysieke activiteiten en mobiliteit
- Emotionele ondersteuning en mentale gezondheid
- Misvattingen rond het Chediak-Higashi-syndroom
- Het Chediak-Higashi-syndroom is hetzelfde als een eenvoudige infectieziekte
- Alleen kinderen worden getroffen door het Chediak-Higashi-syndroom
- Patiënten met het Chediak-Higashi-syndroom hebben geen behandeling nodig
- De symptomen van het Chediak-Higashi-syndroom zijn altijd zichtbaar bij de geboorte
- Het Chediak-Higashi-syndroom heeft altijd een fatale afloop
- Het Chediak-Higashi-syndroom komt alleen voor bij mensen van bepaalde etnische groepen
Synoniemen Chediak-Higashi-syndroom
De gebruikte synoniemen voor het Chediak-Higashi-syndroom zijn:- Begnez-Cesar Syndroom
- Chediak-Steinbrinck-Higashi syndroom
- Leukocytische Anomalie Albinisme Natural Killer lymfocyten
- Defect in Oculocutaan albinisme met leukocytendefect
- Oculocutaan Albinisme, Chediak Higashi-Type
"CHS" is de afkorting van het syndroom.
Epidemiologie aandoening
Het Chediak-Higashi-syndroom is een zeldzame aandoening. Tot nu toe zijn er wereldwijd enkele honderden gevallen gedocumenteerd. Het komt voor bij alle etnische groepen, hoewel sommige studies suggereren dat het iets vaker voorkomt bij mensen van Caucasische afkomst. Mannen en vrouwen worden gelijkmatig getroffen. De symptomen van het syndroom kunnen zich vanaf de geboorte manifesteren, maar in sommige gevallen verschijnen ze pas voor de leeftijd van vijf jaar.Zeldzaamheid en genetische oorzaken
Het syndroom wordt veroorzaakt door mutaties in het LYST-gen en wordt autosomaal recessief overgedragen. Dit betekent dat beide ouders een drager moeten zijn van de genetische mutatie voor de aandoening om zich te ontwikkelen.Geografische en etnische verschillen
Chediak-Higashi-syndroom komt vaker voor bij mensen van Japanse of Inuit-afkomst, hoewel het wereldwijd kan optreden bij mensen van verschillende etnische achtergronden.Mechanisme
Het Chediak-Higashi-syndroom wordt gekarakteriseerd door een defect in de afbraak van ziekteverwekkers door witte bloedcellen, wat het immuunsysteem verzwakt. Dit defect leidt ook tot een verminderde productie van melanine in de huid, wat albinisme veroorzaakt.Leukocytendefect en immuundeficiëntie
Door de defecte functie van lysosomen in witte bloedcellen kunnen deze cellen ziekteverwekkers niet effectief opruimen, waardoor het lichaam vatbaarder wordt voor infecties. Dit kan leiden tot herhaalde infecties van de huid, luchtwegen en andere organen.Albinisme en visuele afwijkingen
Het defect in melanosomen leidt tot een vermindering van het pigment melanine in de huid, haar en ogen, wat resulteert in albinisme. Dit kan ook visuele afwijkingen veroorzaken, zoals nystagmus (oncontroleerbare oogbewegingen) en verminderd zicht.Oorzaken en erfelijkheid
Het Chediak-Higashi-syndroom wordt veroorzaakt door een mutatie in het LYST-gen. Deze aandoening wordt overgeërfd volgens een autosomaal recessieve overervingswijze. Dit betekent dat beide ouders drager moeten zijn van het gemuteerde gen om het syndroom te kunnen doorgeven aan hun kind. Dragers van het mutatie-gen vertonen doorgaans zelf geen symptomen.Risicofactoren
Aangezien Chediak-Higashi-syndroom een erfelijke aandoening is, vormen familieleden van een drager een risicogroep. De aandoening komt voornamelijk voor bij mensen van bepaalde etnische groepen, zoals de Inuit en de Japanse bevolking.Erfelijke aanleg en familiegeschiedenis
Patiënten met een familiegeschiedenis van Chediak-Higashi-syndroom lopen een verhoogd risico. Beide ouders moeten dragers zijn van de genetische mutatie om de aandoening door te geven aan hun kinderen.Etnische prevalentie
Mensen met een etnische achtergrond van Japanse of Inuit afkomst hebben een verhoogd risico op het ontwikkelen van het syndroom vanwege genetische variaties die vaker voorkomen in deze populaties.Risicogroepen
Gezinnen met een genetische geschiedenis van Chediak-Higashi-syndroom, evenals mensen uit bevolkingsgroepen die meer vatbaar zijn, moeten zich bewust zijn van de tekenen en symptomen van de aandoening.Familieleden van dragers
Gezinnen waarvan een van de ouders drager is van de genetische mutatie, moeten zich bewust zijn van het risico van het ontwikkelen van het syndroom bij hun kinderen.Mensen van specifieke etnische afkomst
Mensen van Japanse of Inuit-afkomst hebben een verhoogd risico om de genetische mutatie te dragen en kunnen daarom een verhoogd risico op Chediak-Higashi-syndroom hebben.Symptomen: Albinisme met leukocytendefect
ZuigelingenBij zuigelingen die met het Chediak-Higashi-syndroom worden geboren, kunnen de volgende symptomen optreden: [LIST adenopathie (klieraandoening)
aften (mondzweren)
café-au-lait-vlekken (koffie-met-melk-vlekken op de huid)
ernstige pyodermie (huidaandoening met pusvorming)
geelzucht
gingivitis (ontsteking van het tandvlees door plaque op de tanden)
overmatig zweten (hyperhidrose)
koorts zonder duidelijke oorzaak
miliaria (zweetuitslag)
terugkerende sinus- en longinfecties [/LIST]
Immuunsysteem
Door de aantasting van het immuunsysteem hebben patiënten met het Chediak-Higashi-syndroom een verhoogde gevoeligheid voor virale en bacteriële infecties. Deze infecties zijn vaak ernstig en kunnen levensbedreigend zijn. Het immuunsysteem is verminderd in het bestrijden van indringers, wat leidt tot frequente en hardnekkige infecties vanaf de geboorte of kindertijd.
Oculocutaan albinisme
Een ander belangrijk kenmerk van het Chediak-Higashi-syndroom is oculocutaan albinisme, dat leidt tot een lichte huid, haar en ogen (hypopigmentatie). Patiënten hebben vaak een lichte huid en lichtblond of zilvergrijs haar. Oculocutaan albinisme veroorzaakt ook visuele problemen zoals verminderde gezichtsscherpte, nystagmus (onwillekeurige oogbewegingen) en fotofobie (gevoeligheid voor licht).
Bloedstollingsproblemen
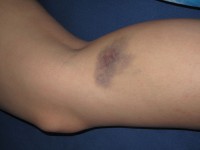 Blauwe plekken zijn kenmerkend /
Blauwe plekken zijn kenmerkend / Veel patiënten met het Chediak-Higashi-syndroom ervaren bloedstollingsproblemen, wat leidt tot gemakkelijk ontstaan van blauwe plekken en abnormaal bloeden. In sommige gevallen kan het syndroom ook het zenuwstelsel aantasten, wat resulteert in zwakte, onhandigheid, moeite met lopen en epileptische aanvallen.
Versnelde fase
Bij ongeveer 85% van de patiënten ontwikkelt zich een versnelde fase, gekarakteriseerd door ongecontroleerde deling van witte bloedcellen en invasie van verschillende organen door deze cellen. Deze fase gaat vaak gepaard met koorts, abnormale bloedingen, ernstige infecties en orgaanfalen. Zonder effectieve behandeling is deze fase levensbedreigend en kan leiden tot vroegtijdig overlijden.
Mildere vorm
Een klein percentage van de patiënten ervaart een mildere vorm van het syndroom, die later in het leven opkomt. Deze vorm gaat vaak gepaard met minder ernstige pigmentveranderingen en minder frequente ernstige infecties. Desondanks kunnen patiënten nog steeds last krijgen van progressieve neurologische problemen zoals tremoren (bevingen), ataxie (bewegings- en evenwichtsstoornissen), perifere neuropathie (zwakte en gevoelloosheid in de armen en benen) en verminderde intellectuele functie.
Alarmsymptomen
Symptomen van het Chediak-Higashi-syndroom kunnen onder andere ernstige herhaalde infecties, vergrote lever en milt, abnormale bloedstolling, en neurologische symptomen zoals vertraagde motorische ontwikkeling omvatten.Herhaalde infecties
Mensen met Chediak-Higashi-syndroom hebben vaak terugkerende infecties, zoals longontsteking, huidinfecties en bloedinfecties. Dit is te wijten aan het verzwakte immuunsysteem.Neurologische en hematologische complicaties
Neurologische problemen, zoals vertraagde ontwikkeling en neurologische achterstand, komen vaak voor, evenals hematologische problemen zoals een verhoogd risico op bloedingen of bloedarmoede door het defect in de witte bloedcellen.Diagnose en onderzoeken
De diagnose van het Chediak-Higashi-syndroom wordt bevestigd door een combinatie van tests, waaronder:- Bloedonderzoek Beenmergbiopsie Oogheelkundig onderzoek Microscopisch onderzoek van de haren
De definitieve diagnose wordt gesteld door genetisch onderzoek naar het LYST-gen.
 Bescherming tegen de zon is nodig / Bron: Dimitrisvetsikas1969, Pixabay
Bescherming tegen de zon is nodig / Bron: Dimitrisvetsikas1969, PixabayBehandeling
Hoewel er geen genezing voor het Chediak-Higashi-syndroom bestaat, richt de behandeling zich op het ondersteunen en verlichten van de symptomen. Vroegtijdige behandeling is cruciaal voor een betere prognose. Behandelopties omvatten:- Beenmergtransplantatie, die in sommige gevallen kan helpen
- Antibiotica om infecties te bestrijden
- Vitamine C-therapie, die kan helpen bij het verbeteren van het immuunsysteem en de bloedstolling
- Gebruik van corrigerende brillen voor visuele problemen
- Bescherming tegen zonlicht met zonnebrandmiddelen, een zonnehoed, zonnebril met een speciale filter en andere beschermende maatregelen
- Chemotherapie en antivirale middelen bij de versnelde fase van de ziekte
Prognose van de aandoening
De prognose van het Chediak-Higashi-syndroom is over het algemeen somber. Veel patiënten overlijden voor de leeftijd van tien jaar door de ernstige symptomen van de versnelde fase. Patiënten bij wie de symptomen later optreden, kunnen gemiddeld tot hun twintigste leven. Zij kunnen echter nog steeds geconfronteerd worden met progressieve neurologische symptomen zoals dementie, perifere neuropathie, parkinsonisme en andere neurologische problemen die hun kwaliteit van leven ernstig beïnvloeden.Complicaties
Bij patiënten met het Chediak-Higashi-syndroom kunnen verschillende complicaties optreden, waaronder:- Versnelde fase van de ziekte met levensbedreigende symptomen
- Chronische infecties door een zwak immuunsysteem
- Neurodegeneratieve problemen zoals tremoren, ataxie en dementie
- Bloedstollingsstoornissen die kunnen leiden tot ernstige bloedingen
Preventie
Er is geen specifieke preventie voor het Chediak-Higashi-syndroom, aangezien het een genetische aandoening is. Het is echter belangrijk om vroegtijdig medische hulp te zoeken bij de eerste symptomen en om een multidisciplinaire behandeling te starten om de kwaliteit van leven te verbeteren en complicaties te minimaliseren.Praktische tips voor het leven met Chediak-Higashi-syndroom
Het Chediak-Higashi-syndroom is een zeldzame genetische aandoening die het immuunsysteem beïnvloedt, wat leidt tot problemen met de afweer tegen infecties, evenals andere gezondheidsproblemen zoals problemen met de ogen, huid, en zenuwen. Het syndroom kan ook invloed hebben op de pigmentatie van de huid en ogen. Het vereist nauwkeurige opvolging en zorg. Hier zijn enkele praktische tips voor het omgaan met Chediak-Higashi-syndroom.Regelmatige controleafspraken en medische opvolging
Vanwege de diverse symptomen van het Chediak-Higashi-syndroom is het belangrijk om regelmatige controleafspraken te hebben bij je arts. Tijdens deze afspraken kunnen bloedonderzoeken worden uitgevoerd om te controleren op infecties of veranderingen in je gezondheid. Het is essentieel om de bloedonderzoeken en andere diagnostische tests bij te houden om complicaties vroegtijdig op te sporen. Het kan ook nodig zijn om je medicatie aan te passen om het immuunsysteem te ondersteunen.Bescherming tegen infecties
Mensen met het Chediak-Higashi-syndroom hebben vaak een verzwakt immuunsysteem, waardoor ze vatbaarder zijn voor infecties. Het is belangrijk om te zorgen voor een strikte hygiëne, zoals het regelmatig wassen van je handen en het vermijden van contact met mensen die ziek zijn. Het dragen van een masker in drukke omgevingen kan helpen om infecties te voorkomen. Zorg ook voor een goed gevaccineerd immuunsysteem door je vaccinatieschema te volgen en te overleggen met je arts over aanvullende vaccinaties.Oogzorg en bescherming tegen licht
Een van de kenmerken van het Chediak-Higashi-syndroom is het effect op de ogen, waarbij er vaak sprake is van visuele problemen zoals photofobie (gevoeligheid voor licht). Het dragen van een zonnebril met UV-bescherming kan helpen om je ogen te beschermen tegen fel licht. Het is ook nuttig om te investeren in vergrotende hulpmiddelen, zoals vergrootglazen of een speciaal vergrootscherm voor digitale apparaten, zodat je gemakkelijker kunt lezen en werken zonder extra stress voor je ogen.Evenwichtig voedingspatroon en vitamine-inname
Het handhaven van een evenwichtig voedingspatroon is essentieel voor het behouden van een goede gezondheid, vooral als je het Chediak-Higashi-syndroom hebt. Voedingsstoffen zoals vitamine C en zink kunnen het immuunsysteem ondersteunen en het lichaam helpen bij het bestrijden van infecties. Overleg met je arts of een diëtist over het nemen van aanvullende vitamines of voedingssupplementen die speciaal gericht zijn op het versterken van je immuunsysteem.Fysieke activiteiten en mobiliteit
Hoewel het Chediak-Higashi-syndroom invloed kan hebben op de zenuwen en mobiliteit, is het nog steeds mogelijk om fysieke activiteiten te doen, zolang je het in je eigen tempo doet. Regelmatige beweging kan helpen om spierkracht en coördinatie te behouden, maar het is belangrijk om overbelasting te voorkomen. Overleg met je arts over geschikte oefeningen en zorg ervoor dat je omgeving veilig is, met antislipmatten en goede verlichting, om valincidenten te vermijden.Emotionele ondersteuning en mentale gezondheid
Het omgaan met de uitdagingen van het Chediak-Higashi-syndroom kan emotioneel belastend zijn. Het is belangrijk om steun te zoeken bij vrienden, familie, of een therapeut. De mentale gezondheid is net zo belangrijk als de fysieke gezondheid, dus zorg ervoor dat je de tijd neemt voor zelfzorg en om te praten over je ervaringen. Er kunnen momenten van frustratie of angst zijn, maar het is belangrijk om hulp te zoeken wanneer dat nodig is.Met de juiste zorg, regelmatige opvolging en een gezonde levensstijl kun je het Chediak-Higashi-syndroom beter beheren en een actieve levensstijl behouden.