Hemofilie: Bloedingsziekte door ontbreken stollingsfactoren
Hemofilie is een zeldzame erfelijke bloedingsaandoening waarbij het bloed niet goed kan stollen. Dit leidt tot langdurige bloedingen na een blessure en, in sommige gevallen, spontane inwendige bloedingen. Inwendige bloedingen kunnen schade aan organen en weefsels veroorzaken en zijn potentieel levensbedreigend. De behandeling van hemofilie omvat substitutietherapie, waarbij de ontbrekende stollingseiwitten worden toegediend. Met de juiste maatregelen kunnen bloedingsproblemen zoveel mogelijk worden voorkomen. De meeste patiënten met hemofilie hebben echter een goede prognose.- Epidemiologie van hemofilie
- Mechanisme
- Oorzaken en erfelijkheid: Ontbreken van stollingsfactoren
- Soorten hemofilie
- Risicofactoren
- Risicogroepen
- Symptomen van hemofilie: Inwendige en uitwendige bloedingen
- Alarmsymptomen
- Diagnose en onderzoeken
- Behandeling van hemofilie
- Prognose van hemofilie
- Complicaties van hemofilie
- Preventie van bloedingsproblemen
- Praktische tips voor het omgaan met hemofilie
- Beheer je behandeling nauwkeurig
- Wees voorbereid op noodgevallen
- Pas je activiteiten aan om blessures te voorkomen
- Houd je gewrichten en spieren gezond
- Herken en behandel bloedingen tijdig
- Zorg voor een evenwichtig voedingspatroon
- Informeer je omgeving over hemofilie
- Misvattingen rond hemofilie
- Hemofilie is een zeldzame aandoening die geen invloed heeft op de dagelijkse activiteiten
- Hemofilie is alleen een probleem voor mannen
- Mensen met hemofilie kunnen niet goed genezen van verwondingen
- Er is geen behandeling voor hemofilie
- Mensen met hemofilie moeten altijd een bloedtransfusie ondergaan bij een bloeding
- Mensen met hemofilie hebben een kortere levensverwachting
Epidemiologie van hemofilie
Hemofilie is een zeldzame erfelijke bloedstollingsstoornis die wereldwijd voorkomt, maar met variërende prevalentie tussen regio's en bevolkingsgroepen. Deze sectie bespreekt de verspreiding, demografie en trends in detail.Prevalentie en incidentie
De geschatte prevalentie van hemofilie A, de meest voorkomende vorm, is ongeveer 1 op de 5.000 mannelijke geboorten, terwijl hemofilie B minder frequent voorkomt, met een prevalentie van ongeveer 1 op de 25.000 mannelijke geboorten. De aandoening is significant zeldzamer bij vrouwen vanwege de X-gebonden overerving.
Regionale variaties
De prevalentie van hemofilie varieert wereldwijd, mede door verschillen in toegang tot diagnose en behandeling. Hogere prevalenties worden vaak gerapporteerd in regio's met geavanceerde gezondheidszorgsystemen, terwijl onderrapportage een probleem blijft in ontwikkelingslanden.
Demografische trends
Hoewel de aandoening voornamelijk mannen treft, kunnen vrouwen die draagsters zijn ook symptomen ontwikkelen, zoals verhoogde bloedingsneiging. Leeftijdsgebonden trends tonen dat levensverwachting en kwaliteit van leven zijn verbeterd door betere behandelingen.
Mechanisme
Hemofilie wordt veroorzaakt door een tekort aan stollingsfactoren, wat leidt tot vertraagde of onvoldoende bloedstolling. Deze sectie bespreekt de pathofysiologie en moleculaire basis van de aandoening.Genetische basis
Hemofilie A wordt veroorzaakt door mutaties in het F8-gen, dat codeert voor stollingsfactor VIII. Hemofilie B is het gevolg van mutaties in het F9-gen, verantwoordelijk voor de productie van factor IX. Beide genen bevinden zich op het X-chromosoom, wat de X-gebonden overerving verklaart.
Stollingscascade en verstoring
In een normale situatie leidt weefselbeschadiging tot activatie van de stollingscascade, waarbij factor VIII en IX een cruciale rol spelen in de vorming van een stevig fibrinenetwerk. Bij hemofiliepatiënten wordt dit proces verstoord, wat resulteert in langdurige bloedingen.
Gevolgen van factordeficiëntie
Het tekort aan stollingsfactoren leidt tot bloedingen in gewrichten, spieren en interne organen. Zonder behandeling kan dit permanente schade veroorzaken, zoals gewrichtsdeformatie en orgaanfalen.
Oorzaken en erfelijkheid: Ontbreken van stollingsfactoren
OorzakenHet lichaam heeft een gespecialiseerd systeem, bekend als de “coagulatiecascade”, dat bloedstolsels vormt door middel van stollingsfactoren (speciale eiwitten). Bij hemofilie ontbreken één of meer van deze stollingsfactoren of functioneren ze niet goed, wat leidt tot een verhoogde bloedingsneiging. Hemofilie wordt voornamelijk veroorzaakt door het ontbreken van stollingsfactor VIII of IX in het bloed. De aandoening is meestal erfelijk en treft vooral mannelijke kinderen. Ongeveer één op de drie patiënten heeft hemofilie zonder bekende familiale geschiedenis.
Erfelijkheid
Hemofilie ontstaat door een geslachtsgebonden (X-chromosomaal) erfelijk stollingsdefect, wat leidt tot een verhoogde bloedingsneiging. Hemofilie komt voornamelijk voor bij mannen. Mannelijke kinderen van een hemofiele vader hebben meestal geen hemofilie, maar dochters van een hemofilie-patiënt kunnen de ziekte doorgeven aan hun zonen. Veranderingen in het F8-gen veroorzaken hemofilie A, terwijl mutaties in het F9-gen Hemofilie B veroorzaken.
Soorten hemofilie
Hemofilie AHemofilie A wordt gekenmerkt door een afwezigheid of tekortkoming van stollingsfactor VIII (antihemofiliefactor). Deze vorm, ook bekend als “klassieke hemofilie” of “factor VIII-deficiëntie”, is de meest voorkomende en komt voor bij ongeveer 80% van de hemofiliepatiënten.
Hemofilie B
Hemofilie B, ook bekend als “kerstziekte” of “factor IX-deficiëntie”, wordt veroorzaakt door het ontbreken van stollingsfactor IX (tromboplastinogeen).
Hemofilie C
Hemofilie C, veroorzaakt door een tekort aan stollingsfactor XI, vertoont vaak milde symptomen. Deze uiterst zeldzame vorm kan zowel mannen als vrouwen treffen.
Risicofactoren
Hemofilie is primair genetisch bepaald, maar andere factoren kunnen de ernst en het verloop van de aandoening beïnvloeden. Deze sectie bespreekt de belangrijkste risicofactoren.Genetische aanleg
De aanwezigheid van een mutatie in het F8- of F9-gen is de belangrijkste risicofactor. Vrouwen met één kopie van de mutatie zijn vaak draagsters en kunnen de aandoening doorgeven aan hun kinderen.
Familiegeschiedenis
Een familiegeschiedenis van hemofilie verhoogt het risico aanzienlijk. Genetisch advies kan nuttig zijn voor families met bekende dragers of patiënten.
Omgevingsfactoren
Hoewel genetica de primaire oorzaak is, kunnen omgevingsfactoren zoals trauma of operaties de frequentie en ernst van bloedingen verhogen. Een proactieve opvolging van deze factoren kan complicaties helpen beperken.
Risicogroepen
Bepaalde groepen patiënten lopen een verhoogd risico op hemofilie of gerelateerde complicaties. Deze sectie bespreekt deze risicogroepen uitgebreid.Mannen met een X-chromosomale mutatie
Mannelijke patiënten zijn het meest vatbaar vanwege de X-gebonden overerving, met een verhoogd risico op ernstige vormen van de aandoening.
Vrouwelijke draagsters
Vrouwen die draagsters zijn, kunnen milde bloedingssymptomen vertonen en hebben een kans van 50% om de mutatie door te geven aan hun kinderen.
Patiënten met beperkte toegang tot zorg
In landen met beperkte toegang tot diagnostiek en behandelingen lopen patiënten een verhoogd risico op complicaties zoals gewrichtsschade en interne bloedingen.
Symptomen van hemofilie: Inwendige en uitwendige bloedingen
Uitwendige bloedingBij hemofilie is de bloedstollingstijd verlengd. Deze symptomen kunnen na de puberteit verbeteren door een stijging van de activiteit van de antihemofiliefactor VIII of IX. Door de verlengde stollingstijd ontwikkelt de patiënt sneller bloedingen (hemorragische diathese) en kan spontane bloeding optreden.
Voorbeelden van symptomen zijn:
- Bloed in de urine of bloed in de ontlasting
- Neusbloedingen zonder duidelijke oorzaak
- Ongewone bloedingen na vaccinaties
- Onverklaarbaar en overmatig bloeden uit snijwonden of verwondingen, of na een operatie of tandheelkundige behandeling
- Onverklaarbare prikkelbaarheid bij zuigelingen
- Pijn, zwelling of een beklemmend gevoel in de gewrichten
- Veel grote of diepe kneuzingen
 Dubbelzien kan een teken zijn van ernstige inwendige bloedingen bij hemofilie / Bron: Frankieleon, Flickr (CC BY-2.0)
Dubbelzien kan een teken zijn van ernstige inwendige bloedingen bij hemofilie / Bron: Frankieleon, Flickr (CC BY-2.0)Hemofiliepatiënten kunnen inwendige bloedingen ontwikkelen op verschillende plaatsen in het lichaam. Bloedingen in de gewrichten (vooral heupen, knieën, enkels en ellebogen) zijn vaak gerapporteerd en kunnen leiden tot zwelling, warmte en pijn. Inwendige bloedingen in de hersenen kunnen ook voorkomen, vooral na een trauma aan het hoofd of een hersenletsel. Kenmerkende symptomen zijn:
- Dubbelzien (diplopie)
- Een acute buik (plotselinge ernstige buikpijn)
- Herhaaldelijk braken
- Langdurige, pijnlijke hoofdpijn of nekpijn of nekstijfheid
- Plotselinge zwakte of onhandigheid van de armen of benen
- Problemen met lopen
- Slaperigheid
- Veranderingen in gedrag
- Stuiptrekkingen (oncontroleerbare fysieke bewegingen en veranderingen in het bewustzijn) of epileptische aanvallen
Alarmsymptomen
Het tijdig herkennen van alarmsymptomen is cruciaal om complicaties bij hemofiliepatiënten te voorkomen. Deze sectie bespreekt de meest voorkomende signalen die medische aandacht vereisen.Spontane bloedingen
Een veelvoorkomend symptoom is spontane bloeding zonder duidelijke oorzaak, vaak in gewrichten (hemartrose), spieren of zachte weefsels.
Langdurige bloedingen na verwondingen
Bloedingen die niet stoppen na kleine verwondingen, zoals snijwonden of tandheelkundige ingrepen, zijn een belangrijk alarmsignaal.
Interne bloedingen
Interne bloedingen in organen zoals het maagdarmkanaal of de hersenen kunnen levensbedreigend zijn en vereisen onmiddellijke medische interventie. Symptomen zoals hevige buikpijn, hoofdpijn of bewustzijnsverlies zijn hierbij indicatief.
Diagnose en onderzoeken
Prenataal onderzoekHemofilie kan prenataal worden gedetecteerd via een vlokkentest, die doorgaans wordt uitgevoerd tussen de tiende en twaalfde zwangerschapsweek.
Lichamelijk en diagnostisch onderzoek
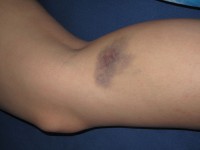 Blauwe plekken zijn kenmerkend voor hemofilie /
Blauwe plekken zijn kenmerkend voor hemofilie / Patiënten met milde symptomen worden vaak pas op latere leeftijd gediagnosticeerd, meestal na een operatie of blessure waarbij overmatig bloeden wordt opgemerkt. Hemofilie wordt vaak vastgesteld wanneer een patiënt plotseling abnormaal begint te bloeden. De arts voert een bloedonderzoek uit om de diagnose te bevestigen. Bij inwendige bloedingen kan de arts bloed in de urine (hematurie), bloed in de ontlasting en grote blauwe plekken opmerken.
Indeling
- Bij milde hemofilie produceert het lichaam 6% tot 50% van de benodigde stollingsfactor. Hierdoor kan de patiënt af en toe langer dan normaal bloeden.
- Bij matige hemofilie produceert het lichaam 2% tot 5% van de stollingsfactor.
- Bij ernstige hemofilie produceert het lichaam minder dan 1% van de stollingsfactor.
Differentiële diagnose
Het bloedonderzoek moet andere aandoeningen uitsluiten die vergelijkbare symptomen veroorzaken, zoals lever- en vaatziekten, gebruik van bepaalde geneesmiddelen en kindermisbruik. Andere aandoeningen om te overwegen zijn het Ehlers-Danlos syndroom (met problemen aan huid en gewrichten), Glanzmann trombasthenie, bloedplaatjesaandoeningen en de ziekte van Von Willebrand.
Behandeling van hemofilie
De meest gebruikelijke behandeling voor hemofilie is de vervangende therapie met stollingsfactoren via intraveneuze infusie. Het is cruciaal dat artsen op de hoogte zijn van de bloedingsstoornis, zodat ze de behandeling kunnen aanpassen aan de behoeften van de patiënt, bijvoorbeeld tijdens een operatie.Prognose van hemofilie
De prognose van hemofilie is sterk verbeterd door moderne behandelingen, maar blijft afhankelijk van de ernst van de aandoening en toegang tot zorg. Deze sectie bespreekt de lange termijnvooruitzichten.Milde tot matige hemofilie
Patiënten met milde vormen hebben doorgaans een goede prognose, mits ze toegang hebben tot preventieve therapieën zoals stollingsfactorconcentraten.
Ernstige hemofilie
Bij ernstige vormen kan de prognose variëren afhankelijk van de frequentie en ernst van bloedingen. Langdurige gewrichtsschade en invaliditeit blijven belangrijke complicaties.
Verbeteringen in levensverwachting
Dankzij profylactische therapieën en verbeterde zorg hebben patiënten nu een bijna normale levensverwachting, vooral in landen met geavanceerde gezondheidszorg.
Complicaties van hemofilie
Mogelijke complicaties bij hemofilie zijn onder andere hemofiliecysten, pseudotumoren die voornamelijk in het bekken en de lange pijpbeenderen voorkomen. Deze cysten kunnen bot vernietigen en weke delen beschadigen. In ernstige gevallen kunnen hersencomplicaties optreden die levensbedreigend kunnen zijn. Andere complicaties zijn diepe inwendige bloedingen met gevoelloosheid en pijn in de ledematen, artritis (gewrichtsontsteking), infecties door bloedtransfusies, en negatieve reacties op de stollingsfactorbehandeling.Preventie van bloedingsproblemen
Het voorkomen van bloedingsproblemen is mogelijk door enkele belangrijke maatregelen:- Gezond gewicht en lichaamsbeweging: Handhaaf een gezond gewicht en zorg voor voldoende lichaamsbeweging om de spieren te versterken en de bloedingsrisico's door verwondingen te verminderen. Zwemmen is een uitstekende keuze voor kinderen met hemofilie omdat het alle spiergroepen gebruikt zonder druk uit te oefenen op de gewrichten.
- Goede tandzorg: Zorg voor goede mondhygiëne om tandvleesbloedingen en andere tandheelkundige problemen te voorkomen.
- Regelmatige medische controle: Volg regelmatig medisch toezicht om bloedingsrisico's te monitoren en behandelingen tijdig aan te passen.
Praktische tips voor het omgaan met hemofilie
Hemofilie is een bloedaandoening waarbij je bloed minder goed stolt. Dit verhoogt het risico op bloedingen, zowel inwendig als uitwendig. Met een zorgvuldige aanpak en de juiste aanpassingen kun je veilig leven en complicaties zoveel mogelijk voorkomen.Beheer je behandeling nauwkeurig
Bij hemofilie is het essentieel om je behandelingsschema goed te volgen. Factorvervangende therapieën zijn vaak de belangrijkste behandeling. Zorg ervoor dat je deze injecties volgens de instructies van je arts toedient of toedieningen op tijd ontvangt in het ziekenhuis. Neem contact op met je arts als je merkt dat de therapie minder effectief lijkt.Wees voorbereid op noodgevallen
Bij hemofilie kunnen kleine verwondingen snel ernstiger worden. Zorg dat je altijd een EHBO-kit bij de hand hebt, inclusief middelen zoals drukverbanden. Je kunt ook een kaart of medisch ID dragen waarop staat dat je hemofilie hebt. Dit kan levensreddend zijn bij een ongeluk of als je een medische behandeling nodig hebt, zoals verdoving of operaties.Pas je activiteiten aan om blessures te voorkomen
Om bloedingen te voorkomen, is het belangrijk om risico's op blessures te minimaliseren. Vermijd sporten met een hoog risico op botsingen of verwondingen, zoals voetbal of boksen. Kies voor veilige alternatieven zoals zwemmen of wandelen, die de belasting op je lichaam beperken en je conditie ondersteunen.Houd je gewrichten en spieren gezond
Inwendige bloedingen bij hemofilie kunnen vaak schade aan gewrichten veroorzaken. Werk samen met een fysiotherapeut om je spieren en gewrichten te versterken, zodat je blessures kunt voorkomen. Regelmatige oefening kan ook helpen om stijfheid en pijn te verminderen.Herken en behandel bloedingen tijdig
Leer de signalen van een inwendige bloeding, zoals zwelling, warmte of pijn in een bepaald gebied. Neem bij twijfel altijd contact op met je arts en start de voorgeschreven behandeling zo snel mogelijk. Het vroeg herkennen van bloedingen kan verdere complicaties helpen voorkomen.Zorg voor een evenwichtig voedingspatroon
Een gezond lichaam ondersteunt een betere bloedcirculatie en helpt complicaties te verminderen. Een evenwichtig voedingspatroon rijk aan mineralen en vitaminen, zoals calcium en vitamine K, kan bijdragen aan het behoud van sterke botten en gezonde bloedvaten. Overleg met een diëtist om ervoor te zorgen dat je de juiste voedingsstoffen binnenkrijgt.Informeer je omgeving over hemofilie
Zorg ervoor dat mensen in je omgeving, zoals familie, vrienden, leraren of collega's, op de hoogte zijn van je aandoening. Dit helpt hen om adequaat te reageren in geval van nood en om je te ondersteunen bij dagelijkse activiteiten. Open communicatie kan ook helpen om stigma te verminderen en begrip te vergroten.Misvattingen rond hemofilie
Hemofilie is een erfelijke aandoening waarbij het bloed niet goed stolt, wat kan leiden tot langdurige bloedingen na verwondingen of zelfs zonder duidelijke oorzaak. Er bestaan veel misvattingen over deze aandoening, vooral over de ernst van de symptomen, de behandeling en het dagelijks leven met hemofilie.Hemofilie is een zeldzame aandoening die geen invloed heeft op de dagelijkse activiteiten
Hoewel hemofilie relatief zeldzaam is, heeft de aandoening wel degelijk invloed op het dagelijks leven van de patiënten. Mensen met hemofilie moeten vaak hun activiteiten aanpassen om risico’s op letsels en bloedingen te vermijden. Sporten die de gewrichten kunnen belasten, zoals hardlopen of contact sporten, moeten vaak vermeden worden, omdat deze de kans op bloedingen in de gewrichten vergroot. De behandeling van hemofilie vereist regelmatige injecties van stollingsfactoren, wat een dagelijkse zorg kan zijn.Hemofilie is alleen een probleem voor mannen
Hoewel het klopt dat hemofilie veel vaker voorkomt bij mannen, kan de aandoening in zeldzame gevallen ook bij vrouwen optreden. Vrouwen hebben twee X-chromosomen, en het gen voor hemofilie zit op het X-chromosoom. In de meeste gevallen worden vrouwen dragers van de aandoening, maar in uitzonderlijke gevallen kunnen zij ook symptomen vertonen als beide X-chromosomen het defecte gen bevatten. Dit komt echter weinig voor.Mensen met hemofilie kunnen niet goed genezen van verwondingen
Het is een misverstand dat mensen met hemofilie nooit goed genezen van verwondingen. Hoewel het bloed stolproces traag kan zijn, kunnen patiënten met hemofilie vaak herstellen van verwondingen en operaties, mits zij tijdig de juiste behandeling krijgen, zoals stollingsfactor-injecties. De juiste medische zorg en behandeling kunnen complicaties voorkomen en het herstel vergemakkelijken. Het is echter belangrijk dat patiënten met hemofilie snel de juiste medische hulp krijgen om bloedingen onder controle te houden.Er is geen behandeling voor hemofilie
Een andere misvatting is dat er geen behandeling beschikbaar is voor hemofilie. In feite bestaan er wel behandelingen die het mogelijk maken om de stollingsfactoren die in het bloed ontbreken, te vervangen. Deze behandelingen kunnen bloedingen helpen voorkomen of stoppen. Patiënten met hemofilie moeten vaak regelmatig stollingsfactoren toegediend krijgen, maar de vooruitgangen in de geneeskunde hebben het voor veel mensen mogelijk gemaakt om een redelijk normaal leven te leiden, ondanks de aandoening.Mensen met hemofilie moeten altijd een bloedtransfusie ondergaan bij een bloeding
Het is niet altijd nodig dat mensen met hemofilie een bloedtransfusie ondergaan bij een bloeding. In veel gevallen kan de bloeding worden gestopt met het toedienen van stollingsfactoren via injecties, zonder dat een volledige bloedtransfusie nodig is. Transfusies kunnen soms wel nodig zijn bij ernstige bloedingen, maar de behandeling met stollingsfactoren is meestal voldoende om de bloeding te beheersen.Mensen met hemofilie hebben een kortere levensverwachting
Dankzij de vooruitgang in de medische behandeling van hemofilie, zoals het regelmatig toedienen van stollingsfactoren, is de levensverwachting van mensen met hemofilie aanzienlijk verbeterd. In het verleden konden bloedingen in de gewrichten en inwendige bloedingen de gezondheid ernstig aantasten, maar moderne behandelingen hebben deze risico’s sterk verminderd. Met de juiste zorg kunnen mensen met hemofilie een lange en actieve levensverwachting hebben.In deze tekst zijn enkele termen die verband houden met medische zorg en behandeling, zoals stollingsfactoren, bloedtransfusies en bloedingen, die kunnen worden opgevolgd met de juiste EHBO zorg en, indien nodig, een behandeling via beeldvormende onderzoeken. Mensen met hemofilie kunnen ook te maken krijgen met complicaties zoals pijn bij bloedingen of gewrichtsproblemen.