Neurofibromatose: Afwijkingen aan huid en ogen
 Neurofibromatose is een erfelijke neurologische aandoening die wordt gekenmerkt door de ontwikkeling van goedaardige tumoren, genaamd neurofibromen, die ontstaan vanuit perifere zenuwen. Deze aandoening komt vaak tot uiting in de adolescentie of jongvolwassenheid en tast voornamelijk de huid aan. De meest opvallende huidafwijkingen zijn café-au-lait-vlekken, die zich kenmerken door hun lichte bruine kleur. Naast de huid kunnen ook de ogen, oogleden en andere delen van het lichaam worden aangetast. Neurofibromatose komt voor in twee hoofdtypes, waarvan de symptomen en ernst sterk variëren. De behandeling richt zich meestal op chirurgie en medicatie, waarbij de vooruitzichten per patiënt verschillen.
Neurofibromatose is een erfelijke neurologische aandoening die wordt gekenmerkt door de ontwikkeling van goedaardige tumoren, genaamd neurofibromen, die ontstaan vanuit perifere zenuwen. Deze aandoening komt vaak tot uiting in de adolescentie of jongvolwassenheid en tast voornamelijk de huid aan. De meest opvallende huidafwijkingen zijn café-au-lait-vlekken, die zich kenmerken door hun lichte bruine kleur. Naast de huid kunnen ook de ogen, oogleden en andere delen van het lichaam worden aangetast. Neurofibromatose komt voor in twee hoofdtypes, waarvan de symptomen en ernst sterk variëren. De behandeling richt zich meestal op chirurgie en medicatie, waarbij de vooruitzichten per patiënt verschillen.
- Epidemiologie van neurofibromatose
- Mechanisme
- Oorzaken en erfelijkheid neurologische aandoening
- Risicofactoren
- Risicogroepen
- Symptomen: Afwijkingen aan Huid en Ogen
- Alarmsymptomen
- Diagnose en onderzoeken
- Behandeling Neurofibromatose met Chirurgie en Medicatie
- Prognose is Variabel
- Complicaties
- Preventie
- Praktische tips voor het leven met neurofibromatose
- Volg een gezond voedingspatroon
- Zorg goed voor jezelf
- Let op veranderingen in je huid en zenuwstelsel
- Zorg voor voldoende beweging en fysieke activiteit
- Werk samen met je zorgteam
- Vermijd overbelasting en stress
- Zorg voor een goede slaaphygiëne
- Houd je mentale gezondheid in de gaten
- Houd je huid in goede conditie
- Zorg voor voldoende ondersteuning bij je dagelijkse activiteiten
- Misvattingen rond neurofibromatose
- Neurofibromatose is altijd ernstig
- Neurofibromatose is een zeldzame aandoening die niemand kent
- Neurofibromatose is altijd erfelijk
- Tumoren bij neurofibromatose zijn altijd gevaarlijk
- Behandeling van neurofibromatose is altijd chirurgisch
- Neurofibromatose leidt altijd tot een verstandelijke beperking
- Neurofibromatose is een aandoening die geen invloed heeft op de levensverwachting
- Neurofibromatose kan worden genezen
Epidemiologie van neurofibromatose
Neurofibromatose is een genetische aandoening die wereldwijd voorkomt en een grote impact heeft op de kwaliteit van leven van de patiënten. De aandoening komt zowel bij mannen als vrouwen voor, en de prevalentie varieert afhankelijk van het type neurofibromatose.Prevalentie van neurofibromatose
De prevalentie van neurofibromatose is wereldwijd relatief constant. Ongeveer 1 op de 3.000 mensen wordt getroffen door neurofibromatose type 1 (NF1), de meest voorkomende vorm van de aandoening. Neurofibromatose type 2 (NF2) komt voor bij ongeveer 1 op de 25.000 mensen. Beide typen komen voor in alle etnische groepen, hoewel er enkele kleine verschillen in prevalentie zijn tussen bevolkingsgroepen.
Erfelijkheid en genetische overerving
Neurofibromatose is meestal autosomaal dominant, wat betekent dat de aandoening van ouder op kind wordt overgedragen. Dit betekent dat een patiënt met neurofibromatose 50% kans heeft om de aandoening aan zijn kinderen door te geven. Ongeveer de helft van de gevallen van NF1 en NF2 komt voort uit een nieuwe mutatie, wat betekent dat geen van de ouders een familiegeschiedenis van de aandoening heeft.
Geografische variaties
De incidentie en prevalentie van neurofibromatose lijken wereldwijd gelijk, hoewel er enkele geografische variaties kunnen zijn in de detectie en diagnose van de aandoening, afhankelijk van de beschikbaarheid van genetisch advies en medische zorg.
Mechanisme
Neurofibromatose is een aandoening die wordt veroorzaakt door mutaties in specifieke genen die de groei en ontwikkeling van zenuwen reguleren. De mechanismen achter de ontwikkeling van neurofibromatose zijn complex en betreffen zowel genetische als moleculaire processen die de celgroei beïnvloeden.Genetische oorzaken
Neurofibromatose type 1 (NF1) wordt veroorzaakt door mutaties in het NF1-gen op chromosoom 17. Dit gen codeert voor neurofibromine, een eiwit dat de groei van zenuwcellen remt. Bij NF1-mutaties ontbreekt neurofibromine, wat leidt tot ongecontroleerde celgroei en de vorming van tumoren, vooral langs de zenuwen.
Neurofibromatose type 2 (NF2) wordt veroorzaakt door mutaties in het NF2-gen op chromosoom 22, dat codeert voor het eiwit merlin. Merlin reguleert de interacties tussen cellen en hun omgeving, en een tekort aan merlin leidt tot de ontwikkeling van tumoren zoals schwannomen.
Tumorontwikkeling en pathologie
De tumoren die ontstaan bij neurofibromatose zijn meestal goedaardig en worden aangeduid als neurofibromen (NF1) of schwannomen (NF2). Deze tumoren groeien uit de schwann-cellen die de zenuwen omhullen, en hoewel ze vaak niet kwaadaardig zijn, kunnen ze ernstige symptomen veroorzaken afhankelijk van hun locatie.
Moleculaire en cellulaire mechanismen
De mechanische basis van de ziekte is ook gerelateerd aan de interactie tussen genetische mutaties en de omgevingsfactoren van de zenuwcellen. De abnormale expressie van genen die coderen voor eiwitten betrokken bij celdeling en groei, zoals neurofibromine en merlin, veroorzaakt tumorvorming en beïnvloedt het normale functioneren van zenuwen en andere weefsels.
Oorzaken en erfelijkheid neurologische aandoening
Neurofibromatose type 1Neurofibromatose type 1 (NF1), ook bekend als de "ziekte van Von Recklinghausen" of "perifere neurofibromatose," is de meest voorkomende vorm van neurofibromatose en komt voor bij ongeveer 95% van alle patiënten met deze aandoening. Het NF1-gen bevindt zich op de lange arm van chromosoom 17. Ongeveer 1 op 3000 personen lijdt aan deze vorm van neurofibromatose.
Neurofibromatose type 2
Neurofibromatose type 2 (NF2), ook wel "akoestische neurofibromatose," "centrale neurofibromatose" of "schwannomatose" genoemd, is een zeldzame erfelijke aandoening die wordt veroorzaakt door mutaties in het NF2-gen, dat zich bevindt op chromosoom 22. Bij NF2 ontwikkelen zich neurofibromen in de hersenen, het ruggenmerg, en meningeomen, wat langzaam groeiende, goedaardige tumoren zijn die ontstaan uit het spinnenwebvlies van de hersenen. Deze vorm komt voor bij ongeveer 1 op 25.000 personen.
Bij ongeveer de helft van de patiënten met neurofibromatose ontstaat de aandoening spontaan, zonder een bekende familiale voorgeschiedenis.
Overerving
Neurofibromatose volgt een autosomaal dominante overervingspatroon, wat betekent dat slechts één kopie van het gemuteerde gen van een ouder voldoende is om de symptomen te veroorzaken.
Risicofactoren
Er zijn verschillende risicofactoren die bijdragen aan de ontwikkeling van neurofibromatose, waarvan genetische factoren de belangrijkste zijn.Genetische erfelijkheid
Zoals eerder vermeld, wordt neurofibromatose meestal overgedragen via een autosomaal dominante erfelijkheidslijn. Dit betekent dat een patiënt een 50% kans heeft om de aandoening aan een kind door te geven. Er is geen manier om de aandoening te voorkomen, maar het wordt vaak gedetecteerd door genetische tests.
Nieuwe genetische mutaties
In sommige gevallen treedt de mutatie van het NF1- of NF2-gen voor het eerst op bij het kind, terwijl geen van de ouders de aandoening heeft. Dit wordt de de novo-mutatie genoemd, en dit gebeurt in ongeveer de helft van de NF1- en NF2-gevallen. Bij deze patiënten is de kans om de aandoening aan hun kinderen door te geven ook 50%.
Omgevingsfactoren
Hoewel neurofibromatose voornamelijk een genetische aandoening is, kunnen omgevingsfactoren een rol spelen in het verergeren van de symptomen of het versnellen van de tumorontwikkeling. Er is echter geen bewezen omgevingsfactor die specifiek neurofibromatose veroorzaakt.
Risicogroepen
Er zijn verschillende groepen die een verhoogd risico lopen op het ontwikkelen van neurofibromatose, vooral op basis van erfelijkheid en familiegeschiedenis.Familieleden van patiënten met neurofibromatose
Familieleden van iemand met neurofibromatose lopen een verhoogd risico op het ontwikkelen van de aandoening. Dit geldt vooral voor kinderen van ouders die de aandoening hebben, aangezien de aandoening autosomaal dominant wordt overgedragen.
Mensen met een de novo-mutatie
Patiënten die een de novo-mutatie hebben, ontwikkelen de aandoening zonder dat er een familiale voorgeschiedenis is. Hoewel deze patiënten zelf geen familiegeschiedenis hebben, is er een kans dat hun kinderen de aandoening erven.
Patiënten met specifieke genetische variaties
Niet iedereen met een genetische aanleg voor neurofibromatose ontwikkelt symptomen. Bij sommige mensen kunnen specifieke genetische variaties de ernst of het type tumoren beïnvloeden dat zich ontwikkelt. Er zijn aanwijzingen dat mutaties in bepaalde regio's van het NF1-gen invloed kunnen hebben op het type en de ernst van de tumoren.
Symptomen: Afwijkingen aan Huid en Ogen
De symptomen van neurofibromatose kunnen sterk variëren, zowel in aard als in ernst, van mild tot ernstig.Type I
Hart
Cardiovasculaire complicaties bij NF1 omvatten aangeboren hartafwijkingen, hypertensie (hoge bloeddruk) en vaatproblemen zoals vernauwingen, blokkades of beschadigde bloedvaten (vasculopathie).
Huid
Patiënten met NF1 ontwikkelen vanaf de geboorte of in de eerste levensjaren vaak zes of meer onregelmatig gevormde café-au-lait-vlekken op de huid. Deze vlekken zijn donkerder dan de omringende huid en meten meer dan 5 mm in diameter. Ze bevinden zich vaak onder de oksels, in de elleboog- en knieholtes, en rond de liesstreek. De vlekken nemen toe in grootte en aantal gedurende de eerste tien levensjaren. Daarnaast ontwikkelen zich op of onder de huid knobbeltjes genaamd neurofibromen, die op elke leeftijd kunnen verschijnen. Vaak zijn er meerdere neurofibromen aanwezig, en soms groeien deze verder in het lichaam.
Ogen
Bij NF1 kunnen de oogleden verdikken en onregelmatig van vorm worden door de groei van neurofibromen. Dit kan leiden tot verhoogde oogboldruk (glaucoom) of een lui oog (amblyopie). Op de iris (regenboogvlies) ontstaan pigmentvlekjes, bekend als Lisch-knobbeltjes, die meestal geen invloed hebben op het gezichtsvermogen. Een ander mogelijk gevolg is een goedaardige tumor op de oogzenuw, een opticusglioom, die wel visusproblemen kan veroorzaken.
Andere Symptomen
NF1 kan ook de botontwikkeling beïnvloeden, wat kan leiden tot skeletafwijkingen zoals scoliose, afwijkingen aan de armen en benen, en een groot hoofd (macrocefalie). Andere mogelijke symptomen zijn hoofdpijn, epileptische aanvallen, verminderde intellectuele functies, eenzijdig gehoorverlies, ontwikkelingsstoornissen en kort gestalte. In zeldzame gevallen kan NF1 leiden tot kanker zoals leukemie, Wilmstumor, rhabdomyosarcoom en feochromocytoom.
Type II
NF2 komt vaak tot uiting in de tienerjaren of jongvolwassenheid met gehoorproblemen veroorzaakt door een tumor op de gehoorzenuw, een akoestisch neuroom. Andere symptomen zijn hoofdpijn, evenwichtsproblemen, oorsuizen, vroege ontwikkeling van cataract (posterior subcapsulaire cataract), Lisch-knobbeltjes, en zwakte in een arm of been.
Alarmsymptomen
Het herkennen van alarmsymptomen is cruciaal voor een vroege diagnose van neurofibromatose. De symptomen kunnen variëren, afhankelijk van het type neurofibromatose en de betrokken zenuwen.Opkomst van gezwellen
De meest kenmerkende symptomen van neurofibromatose zijn de verschijnen van gezwellen of knobbels langs zenuwen. Bij NF1 ontstaan vaak neurofibromen op de huid, terwijl NF2 vaak gekarakteriseerd wordt door tumoren in de hersenen, zoals schwannomen op de gehoorzenuwen.
Verlies van gehoor en evenwicht
Bij patiënten met neurofibromatose type 2 kunnen tumoren op de gehoorzenuwen leiden tot gehoorverlies, evenwichtsstoornissen en duizeligheid. Dit kan progressief zijn en leidt vaak tot permanente schade aan het gehoor.
Veranderingen in huidkleur of andere zichtbare tekenen
Bij NF1 kunnen patiënten vaak café-au-lait vlekken op de huid ontwikkelen, die bruine vlekken zijn die kenmerkend zijn voor de aandoening. Dit is een van de vroege tekenen die artsen helpen bij de diagnose.
Diagnose en onderzoeken
De diagnose van neurofibromatose wordt gesteld door een combinatie van lichamelijk onderzoek, dermatologisch onderzoek, gehooronderzoek, oogonderzoek en neurologisch onderzoek. Beeldvormende technieken zoals röntgenfoto's, MRI-scans en CT-scans helpen bij het opsporen van interne tumoren en andere afwijkingen. Genetisch onderzoek kan de diagnose bevestigen, wat belangrijk is voor het uitsluiten van andere aandoeningen zoals het Legius-syndroom of het Noonan-syndroom.Behandeling Neurofibromatose met Chirurgie en Medicatie
De behandeling van neurofibromatose richt zich voornamelijk op symptoombeheer. Ooglidproblemen kunnen chirurgisch worden behandeld, hoewel de kans op terugkeer van de neurofibromen groot is. Behandeling van opticusgliomen is meestal niet nodig als het gezichtsvermogen niet aangetast is. Chirurgische verwijdering van tumoren is mogelijk, vooral als ze de hersenen bedreigen, waarbij soms ook chemotherapie en radiotherapie nodig zijn. Medicatie kan helpen bij het behandelen van hoofdpijn en epilepsie. Bij ernstige gehoorproblemen kan een cochleair implantaat een oplossing bieden.Prognose is Variabel
NF1NF1 is een progressieve aandoening, wat betekent dat de symptomen na verloop van tijd kunnen verergeren. Bij sommige patiënten blijven de symptomen echter stabiel. De ernst en het verloop van de ziekte variëren sterk tussen patiënten. De levensverwachting is meestal normaal, hoewel de aanwezigheid van neurofibromen kan leiden tot cosmetische en psychologische problemen.
NF2
NF2 is zeldzamer dan NF1 en de symptomen zijn vaak ernstiger. De ontwikkeling van brughoektumoren kan leiden tot progressieve gehoor- en evenwichtsproblemen. Jongere patiënten met meningeomen hebben doorgaans een slechtere prognose dan oudere patiënten.
Complicaties
Mogelijke complicaties zijn:- aantasting van het uiterlijk
- ademhalingsproblemen
- cardiovasculaire problemen
- een goedaardige bijniertumor (feochromocytoom)
- gedeeltelijke of volledige doofheid
- gewrichtsaandoeningen
- hormonale veranderingen
- kanker (zoals borstkanker, leukemie, hersentumoren en sommige vormen van weke delen kanker)
- kleine goedaardige huidtumoren (huid schwannoma's)
- meerdere goedaardige hersentumoren of spinale tumoren
- neurologische problemen (ontwikkelingsstoornissen, epilepsie, hydrocefalie, )
- problemen met het gezichtsvermogen
- zenuwbeschadiging aan het gezicht
- zwakte of gevoelloosheid in de armen en/of benen en/of het gezicht (gezichtszwakte)
Preventie
Aangezien neurofibromatose meestal genetisch wordt doorgegeven, is er geen manier om de aandoening volledig te voorkomen. Er zijn echter manieren om de symptomen te beheren en de impact op het leven van de patiënt te verminderen.Genetisch advies voor familieleden
Gezinsleden van mensen met neurofibromatose kunnen baat hebben bij genetisch advies om hun eigen risico te begrijpen en te onderzoeken of ze draagsters van de aandoening zijn. Dit is vooral belangrijk voor mensen die overwegen kinderen te krijgen.
Vroegtijdige diagnose en monitoring
Vroege diagnose kan helpen bij het identificeren van tumoren en het starten van behandelingen die symptomen kunnen verlichten en de progressie van de aandoening kunnen vertragen. Regelmatige controles en screenings kunnen de diagnose versnellen.
Beheer van symptomen
Hoewel er geen manier is om neurofibromatose volledig te genezen, kan het beheer van symptomen zoals pijn, gehoorverlies en andere neurologische complicaties de levenskwaliteit van patiënten verbeteren.
Praktische tips voor het leven met neurofibromatose
Volg een gezond voedingspatroon
Het behouden van een gezond en evenwichtig voedingspatroon is essentieel voor het beheer van neurofibromatose. Voeding speelt een cruciale rol in je algemene gezondheid en kan je helpen om het risico op complicaties te verlagen. Zorg ervoor dat je voldoende vitamines en mineralen binnenkrijgt, met name vitamine A, C en D, die bijdragen aan het behoud van een gezonde huid en het versterken van het immuunsysteem. Overweeg om gezonde vetten, zoals die in avocados, olijfolie, en noten, toe te voegen aan je dieet, omdat ze ontstekingsremmende eigenschappen hebben die kunnen helpen bij het beheren van de aandoening.Zorg goed voor jezelf
Naast een gezond voedingspatroon is het belangrijk om goed voor jezelf te zorgen als je leeft met neurofibromatose. Regelmatige medische controles zijn essentieel om de voortgang van de aandoening te volgen en om mogelijke complicaties vroegtijdig te identificeren. Dit kan onder meer regelmatige beeldvormende onderzoeken inhouden om tumoren of veranderingen in je zenuwstelsel te monitoren. Houd ook rekening met je fysieke en mentale welzijn: zorg voor voldoende rust, vermijd stress en zoek steun bij een therapeut als je mentale gezondheid een uitdaging vormt.Let op veranderingen in je huid en zenuwstelsel
Een belangrijk aspect van neurofibromatose is het effect op je huid en zenuwstelsel, dus wees alert op veranderingen. Controleer regelmatig op nieuwe huidziekten of veranderingen in bestaande huidtumoren, zoals neurofibromen. Als je nieuwe knobbels of andere veranderingen opmerkt, informeer dan je arts. Dit is belangrijk voor het snel identificeren van tumoren die mogelijk groei vertonen. Symptomen zoals pijn, gevoelloosheid, of spierzwakte kunnen duiden op zenuwproblemen, dus wees alert en bespreek deze met je arts.Zorg voor voldoende beweging en fysieke activiteit
Fysieke activiteit kan helpen om je spieren sterk en flexibel te houden, wat essentieel is, vooral als neurofibromatose invloed heeft op je bewegingsvermogen. Beweging kan ook bijdragen aan je algehele gezondheid en het verminderen van stress. Zorg ervoor dat je een activiteit kiest die bij je past, zoals wandelen, zwemmen of yoga. Als je moeite hebt met bepaalde bewegingen, kan fysiotherapie je helpen bij het verbeteren van je mobiliteit en het verlichten van pijn.Werk samen met je zorgteam
Het leven met neurofibromatose vereist vaak een team van zorgverleners. Regelmatige controle bij een neuroloog, dermatoloog, en andere specialisten is essentieel om de voortgang van de aandoening te monitoren. Bespreek je symptomen en zorgen altijd met je arts. Het is ook belangrijk om openlijk te communiceren over eventuele veranderingen die je in je lichaam of geest ervaart, zodat je zorgteam de juiste ondersteuning kan bieden.Vermijd overbelasting en stress
Chronische aandoeningen zoals neurofibromatose kunnen zowel fysiek als mentaal belastend zijn. Het is belangrijk om jezelf niet te overbelasten en tijd te nemen voor ontspanning. Stress kan de symptomen verergeren, dus zorg ervoor dat je regelmatig ontspanningstechnieken toepast, zoals ademhalingsoefeningen, meditatie, of een rustige wandeling. Je welzijn komt op de eerste plaats, dus wees niet bang om grenzen te stellen en voor jezelf te zorgen.Zorg voor een goede slaaphygiëne
Een goede nachtrust is cruciaal voor je algehele gezondheid. Voor patiënten met neurofibromatose kan een verstoorde slaap het herstel belemmeren en de symptomen verergeren. Zorg ervoor dat je een rustige en comfortabele slaapomgeving hebt en houd een regelmatig slaapschema aan. Het vermijden van schermen voor het slapen gaan en het creëren van een rustgevende sfeer kan je helpen om beter te slapen en je energieker te voelen.Houd je mentale gezondheid in de gaten
Neurofibromatose kan een invloed hebben op je mentaal welzijn. Het omgaan met de fysieke aspecten van de aandoening kan emotioneel zwaar zijn. Het is belangrijk om tijd te nemen voor je mentale gezondheid. Zoek steun bij familie, vrienden, of een therapeut om te praten over eventuele zorgen of emoties die je hebt. Als je merkt dat stress, angst, of depressie een rol speelt, aarzel dan niet om professionele hulp te zoeken. Je mentale gezondheid speelt een cruciale rol in je algehele herstel en welzijn. Mentale gezondheid kan net zo belangrijk zijn als het beheren van je fysieke symptomen.Houd je huid in goede conditie
Patiënten met neurofibromatose kunnen een verhoogd risico hebben op huidafwijkingen. Het is belangrijk om je huid goed te verzorgen en regelmatig te controleren op veranderingen, zoals nieuwe huidziekten of tumoren. Gebruik milde huidverzorgingsproducten en bescherm je huid tegen overmatige zonblootstelling. Als je huidproblemen of nieuwe gezwellen opmerkt, raadpleeg dan onmiddellijk je arts voor advies.Zorg voor voldoende ondersteuning bij je dagelijkse activiteiten
Neurofibromatose kan invloed hebben op je vermogen om bepaalde dagelijkse taken uit te voeren. Het kan nuttig zijn om ondersteuning te zoeken bij een ergotherapeut om je te helpen bij het aanpassen van je omgeving. Denk aan hulpmiddelen voor mobiliteit of strategieën voor het uitvoeren van taken die anders moeilijk zouden zijn. Vraag ook familieleden of vrienden om hulp bij het uitvoeren van taken die fysiek belastend kunnen zijn.Misvattingen rond neurofibromatose
Neurofibromatose is een genetische aandoening die tumoren veroorzaakt op zenuwweefsel, vaak in de vorm van goedaardige tumoren, de zogenaamde neurofibromen. Deze tumoren kunnen zich overal in het lichaam vormen, maar zijn het vaakst te vinden op of nabij de huid. Er bestaan verschillende vormen van neurofibromatose, waaronder neurofibromatose type 1 (NF1) en neurofibromatose type 2 (NF2). Er heersen echter veel misvattingen over de aandoening, de symptomen en de mogelijke behandeling ervan. In dit artikel worden enkele van de meest voorkomende misvattingen besproken.Neurofibromatose is altijd ernstig
Veel mensen denken dat neurofibromatose altijd ernstige gezondheidsproblemen met zich meebrengt, maar dit is niet het geval. De ernst van de aandoening varieert sterk, afhankelijk van het type neurofibromatose en de specifieke symptomen die zich voordoen. In veel gevallen veroorzaakt de aandoening geen ernstige problemen en zijn de tumoren goedaardig. Sommige mensen ervaren geen symptomen of slechts milde symptomen die geen invloed hebben op hun dagelijks leven. In andere gevallen kunnen de tumoren echter pijn veroorzaken, het gezichtsvermogen beïnvloeden of andere complicaties veroorzaken, afhankelijk van hun locatie in het lichaam.Neurofibromatose is een zeldzame aandoening die niemand kent
Hoewel neurofibromatose misschien minder bekend is dan andere genetische aandoeningen, komt het veel vaker voor dan vaak wordt gedacht. Neurofibromatose type 1 (NF1) is een van de meest voorkomende genetische aandoeningen en komt bij ongeveer 1 op de 3.000 mensen voor. De aandoening kan in alle etnische groepen voorkomen en wordt vaak al op jonge leeftijd gediagnosticeerd. Neurofibromatose is dus geen zeldzame aandoening, maar er is relatief weinig publieke bewustwording over de aandoening.Neurofibromatose is altijd erfelijk
Neurofibromatose kan inderdaad erfelijk zijn, maar het is niet altijd het geval. In veel gevallen ontstaat de aandoening als gevolg van een spontane mutatie in een gen, wat betekent dat het kan optreden zonder dat er een familiegeschiedenis van neurofibromatose is. Wanneer de aandoening erfelijk is, wordt deze meestal op een autosomaal dominante manier doorgegeven, wat betekent dat een kind van een ouder met neurofibromatose 50% kans heeft om de aandoening te erven. Echter, in gevallen van spontane mutaties is er geen familiegeschiedenis van de aandoening.Tumoren bij neurofibromatose zijn altijd gevaarlijk
Een van de grootste misvattingen over neurofibromatose is dat de tumoren altijd gevaarlijk zijn. In werkelijkheid zijn de meeste tumoren die worden geassocieerd met neurofibromatose goedaardig en veroorzaken ze geen ernstige problemen. De tumoren kunnen echter wel pijn of ongemak veroorzaken, afhankelijk van hun grootte en locatie. In zeldzame gevallen kunnen de tumoren zich ontwikkelen tot kwaadaardige vormen, zoals neurofibrosarcomen, maar dit komt slechts in een klein percentage van de gevallen voor. Regelmatige medische controles en beeldvormende onderzoeken kunnen helpen bij het monitoren van de tumoren en het identificeren van eventuele risicofactoren.Behandeling van neurofibromatose is altijd chirurgisch
Chirurgie is een mogelijke behandelingsoptie voor neurofibromatose, vooral als de tumoren pijn veroorzaken of de functie van nabijgelegen organen beïnvloeden. Echter, niet alle gevallen van neurofibromatose vereisen chirurgische ingrepen. Bij veel patiënten worden de tumoren gecontroleerd zonder dat een operatie noodzakelijk is. In plaats daarvan kan een arts besluiten om de tumoren te monitoren door middel van regelmatige controles en beeldvormende onderzoeken, vooral als de tumoren geen symptomen veroorzaken. In sommige gevallen kunnen andere behandelingen, zoals medicatie of bestraling, ook worden overwogen.Neurofibromatose leidt altijd tot een verstandelijke beperking
Hoewel neurofibromatose geassocieerd kan worden met leermoeilijkheden, vooral bij mensen met NF1, leidt het niet noodzakelijk tot een verstandelijke beperking. De meeste mensen met neurofibromatose hebben een normale intelligentie en kunnen een volwaardig leven leiden. Echter, sommige mensen met NF1 kunnen moeite hebben met bepaalde cognitieve functies, zoals aandacht en geheugen. De mate van invloed op het intellectuele functioneren varieert sterk tussen individuen. Bij neurofibromatose type 2 (NF2) komen cognitieve problemen veel minder voor. Het is belangrijk om te begrijpen dat neurofibromatose in veel gevallen geen grote invloed heeft op de intellectuele vermogens van de patiënt.Neurofibromatose is een aandoening die geen invloed heeft op de levensverwachting
In de meeste gevallen heeft neurofibromatose geen invloed op de levensverwachting van de patiënt, vooral als de tumoren goedaardig blijven en geen ernstige complicaties veroorzaken. Echter, in sommige gevallen kunnen de tumoren zich ontwikkelen tot kwaadaardige tumoren, wat kan leiden tot gezondheidsproblemen die de levensverwachting beïnvloeden. Het is belangrijk om te benadrukken dat de meeste mensen met neurofibromatose een normaal leven kunnen leiden met de juiste medische zorg en opvolging, en dat de aandoening zelf zelden levensbedreigend is.Neurofibromatose kan worden genezen
Er is momenteel geen genezing voor neurofibromatose, maar de aandoening kan in veel gevallen goed worden beheerd. Behandeling is vaak gericht op het verlichten van symptomen en het voorkomen van complicaties. Regelmatige medische controles, beeldvormende onderzoeken, en ondersteuning van gespecialiseerde zorgverleners kunnen patiënten helpen om de aandoening te beheren. Hoewel er geen genezing is, kunnen mensen met neurofibromatose een relatief normaal en gezond leven leiden met de juiste behandeling en zorg.© 2016 - 2025 Miske, het auteursrecht van dit artikel ligt bij de infoteur. Zonder toestemming is vermenigvuldiging verboden. Deze informatie is van informatieve aard en geen vervanging voor professioneel medisch advies. Per 2021 gaat InfoNu verder als archief, artikelen worden nog maar beperkt geactualiseerd.
 Café au lait vlekken: symptomen, oorzaak en verwijderenCafé-au-lait is een koffie verkeerd op z'n Frans. Een café au lait macula (CALM) of café au lait vlekken is een lichtbru…
Café au lait vlekken: symptomen, oorzaak en verwijderenCafé-au-lait is een koffie verkeerd op z'n Frans. Een café au lait macula (CALM) of café au lait vlekken is een lichtbru…
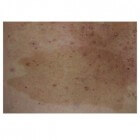 Café-au-lait-vlekken: Koffie-met-melk-vlekken op huidCafé-au-lait-vlekken zijn lichtbruine tot donkerbruine huidvlekken die overal op de huid kunnen verschijnen. Ze zijn aan…
Café-au-lait-vlekken: Koffie-met-melk-vlekken op huidCafé-au-lait-vlekken zijn lichtbruine tot donkerbruine huidvlekken die overal op de huid kunnen verschijnen. Ze zijn aan…
 Neurofibromatose type 1; de Ziekte van Von RecklinghausenNeurofibromatose (NF) is redelijk zeldzame, erfelijke aandoening en komt zowel voor bij kinderen als volwassenen. Neurof…
Neurofibromatose type 1; de Ziekte van Von RecklinghausenNeurofibromatose (NF) is redelijk zeldzame, erfelijke aandoening en komt zowel voor bij kinderen als volwassenen. Neurof…
 Moedervlekken: Soorten, vormen, symptomen en behandelingEen moedervlek, in medische termen gekend als een naevus is een soort vlek op de huid die bij de geboorte aanwezig is.…
Moedervlekken: Soorten, vormen, symptomen en behandelingEen moedervlek, in medische termen gekend als een naevus is een soort vlek op de huid die bij de geboorte aanwezig is.…
 Hyperandrogenisme: hormonale afwijking bij vrouwenHyperandrogenisme is een hormonale afwijking waarbij vrouwen te veel androgenen aanmaken. Dit kan leiden tot een hoge te…
Hyperandrogenisme: hormonale afwijking bij vrouwenHyperandrogenisme is een hormonale afwijking waarbij vrouwen te veel androgenen aanmaken. Dit kan leiden tot een hoge te…
 Jeuk in de keel of kriebel in de keel: oorzaken en symptomenJeuk in je keel of mond (gehemelte) of een kriebelende keel, is vaak een klacht die heel erg op de voorgrond treedt. Jeu…
Jeuk in de keel of kriebel in de keel: oorzaken en symptomenJeuk in je keel of mond (gehemelte) of een kriebelende keel, is vaak een klacht die heel erg op de voorgrond treedt. Jeu…
Gerelateerde artikelen
Bronnen en referenties
- Coëlho, Medisch Zakwoordenboek, digitale editie, versie 2010, geraadpleegd op 12 september 2016
- Diagnosis, http://www.mayoclinic.org/diseases-conditions/neurofibromatosis/diagnosis-treatment/diagnosis/dxc-20167903, geraadpleegd op 12 september 2016
- Neurofibromatosis Fact Sheet, http://www.ninds.nih.gov/disorders/neurofibromatosis/detail_neurofibromatosis.htm, geraadpleegd op 12 september 2016
- Neurofibromatosis, https://medlineplus.gov/neurofibromatosis.html, geraadpleegd op 12 september 2016
- Neurofibromatosis, https://www.aapos.org/terms/conditions/79, geraadpleegd op 12 september 2016
- Overview, http://www.mayoclinic.org/diseases-conditions/neurofibromatosis/home/ovc-20167893, geraadpleegd op 12 september 2016
- Symptoms & Causes, http://www.mayoclinic.org/diseases-conditions/neurofibromatosis/symptoms-causes/dxc-20167896, geraadpleegd op 12 september 2016
- Treatment, http://www.mayoclinic.org/diseases-conditions/neurofibromatosis/diagnosis-treatment/treatment/txc-20167907, geraadpleegd op 12 september 2016
Miske (4.039 artikelen)
Laatste update: 23-02-2025
Rubriek: Mens en Gezondheid
Subrubriek: Aandoeningen
Bronnen en referenties: 8
Laatste update: 23-02-2025
Rubriek: Mens en Gezondheid
Subrubriek: Aandoeningen
Bronnen en referenties: 8
Per 2021 gaat InfoNu verder als archief. Het grote aanbod van artikelen blijft beschikbaar maar er worden geen nieuwe artikelen meer gepubliceerd en nog maar beperkt geactualiseerd, daardoor kunnen artikelen op bepaalde punten verouderd zijn. Reacties plaatsen bij artikelen is niet meer mogelijk.
Medische informatie…
Deze informatie is van informatieve aard en geen vervanging voor professioneel medisch advies. Raadpleeg bij medische problemen en/of vragen altijd een arts.
Deze informatie is van informatieve aard en geen vervanging voor professioneel medisch advies. Raadpleeg bij medische problemen en/of vragen altijd een arts.