Apert-syndroom: Craniofaciale afwijking
 Het Apert-syndroom is een genetische aandoening waarbij de naden van de schedelbeenderen eerder sluiten dan normaal (craniosynostose). De schedelontwikkeling verloopt hierdoor abnormaal, waardoor de vorm van het hoofd en het gezicht afwijkend is. Ook de fusie van vingers en tenen kan voorkomen. Daarnaast zijn tal van bijkomende symptomen gerapporteerd. Het Apert-syndroom is vernoemd naar de Franse arts E. Apert die het syndroom voor het eerst beschreef in 1906.
Het Apert-syndroom is een genetische aandoening waarbij de naden van de schedelbeenderen eerder sluiten dan normaal (craniosynostose). De schedelontwikkeling verloopt hierdoor abnormaal, waardoor de vorm van het hoofd en het gezicht afwijkend is. Ook de fusie van vingers en tenen kan voorkomen. Daarnaast zijn tal van bijkomende symptomen gerapporteerd. Het Apert-syndroom is vernoemd naar de Franse arts E. Apert die het syndroom voor het eerst beschreef in 1906.
- Synoniemen Apert-syndroom
- Epidemiologie craniofaciale afwijking
- Mechanisme
- Genetische basis
- Effect op organen en functies
- Oorzaken en erfelijkheid aandoening
- Symptomen: Gezicht, ogen, vingers en tenen
- Diagnose en onderzoeken
- Behandeling
- Prognose aandoening
- Complicaties
- Preventie
- Praktische tips voor het leven met Apert-syndroom
- Volg een gezonde levensstijl
- Zorg voor je fysieke gezondheid en mobiliteit
- Neem contact op met specialisten voor chirurgische ingrepen
- Let op je mentale gezondheid
- Volg een zorgplan voor regelmatige medische controleafspraken
- Misvattingen rond Apert-syndroom
- Apert-syndroom is altijd erfelijk
- Alle patiënten met Apert-syndroom hebben eenzelfde uiterlijk
- Apert-syndroom beïnvloedt enkel de botstructuur
- Alle patiënten met Apert-syndroom hebben een verstandelijke beperking
- Er is geen behandeling mogelijk voor Apert-syndroom
- Patiënten met Apert-syndroom kunnen geen zelfstandig leven leiden
Synoniemen Apert-syndroom
Het Apert-syndroom (ACS I) is eveneens gekend onder deze synoniemen:- Acrocephalosyndactylie (Apert)
- Acrocephalosyndactylie, Type I
- Syndactylische Oxycefalie
- Syndroom van Apert
- Ziekte van Apert
Epidemiologie craniofaciale afwijking
Het Apert-syndroom treft naar schatting 1 op de 65.000 tot 200.000 pasgeborenen wereldwijd. De grote verschillen in de prevalentiecijfers zijn te wijten aan de verschillende geraadpleegde bronnen. In Nederland worden jaarlijks gemiddeld twee babys geboren met het syndroom. Verder is de prevalentie hoger bij Aziaten en lager bij Iberiërs. Het syndroom heeft geen seksuele voorkeur; mannen en vrouwen worden in gelijke aantallen getroffen. Het syndroom is aangeboren, waardoor de symptomen zich al bij zuigelingen presenteren.Mechanisme
Genetische basis
Het Apert-syndroom wordt veroorzaakt door mutaties in het FGFR2-gen (fibroblastgroeifactorreceptor 2), wat leidt tot abnormale botgroei en voortijdige sluiting van de schedelnaden (craniosynostose).Effect op organen en functies
Naast craniosynostose kunnen de mutaties leiden tot syndactylie (samengroeiing van vingers of tenen), ademhalingsproblemen door afwijkingen in het gezichtsskelet en mogelijke ontwikkelingsstoornissen.Oorzaken en erfelijkheid aandoening
Een genmutatie van het FGFR2-gen veroorzaakt het Apert-syndroom. De overerving van deze aandoening verloopt via autosomaal dominante wijze, waardoor één kopie van het gewijzigde gen voldoende is om de aandoening te veroorzaken. Bij bijna alle patiënten met het Apert-syndroom is de ziekte het gevolg van nieuwe mutaties in het gen (de novo-mutatie). Hierdoor is er vaak geen familiale geschiedenis van het syndroom bekend. Patiënten met het Apert-syndroom kunnen echter mogelijk de ziekte doorgeven aan de volgende generatie.Symptomen: Gezicht, ogen, vingers en tenen
Het syndroom van Apert is een multisystemische aandoening waarbij de patiënt voornamelijk problemen heeft met het gezicht, de ogen, en de vingers en tenen. Daarnaast kunnen tal van andere symptomen optreden. De ernst, uitgebreidheid en het optreden van de symptomen variëren per patiënt, zelfs binnen dezelfde familie. De symptomen treden op bij de geboorte.Gezicht
Door de voortijdige fusie van de schedelbotten heeft de patiënt zeer karakteristieke gelaatstrekken. Het hoofd is niet in staat om normaal te groeien, wat leidt tot een gezonken verschijning in het midden van het gezicht. Het hoofd is verder lang met een hoog voorhoofd. De neus heeft een brede, korte en bolvormige tip, waardoor deze er snavelvormig uitziet. De bovenkaak is bovendien onderontwikkeld. Dit resulteert in overvolle tanden en andere gebitsproblemen. Vroege fusie van de schedelbeenderen beïnvloedt ook de ontwikkeling van de hersenen, wat kan leiden tot een verstoorde intellectuele ontwikkeling. De cognitieve vaardigheden bij patiënten met het syndroom van Apert variëren van normaal tot een lichte of matige verstandelijke beperking.
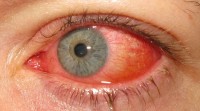 Rode ogen bij conjunctivitis / Bron: Marco Mayer, Wikimedia Commons (CC BY-SA-4.0)
Rode ogen bij conjunctivitis / Bron: Marco Mayer, Wikimedia Commons (CC BY-SA-4.0)De patiënt heeft tevens uitpuilende ogen (exoftalmie) die breed uit elkaar geplaatst zijn (de medische term hiervoor is "hypertelorisme"). Hierbij kunnen soms hoornvliesletsels als gevolg van conjunctivitis, keratitis, en droge ogen voorkomen. Strabisme, amblyopie, optische atrofie, luxatie van de oogbol, keratoconus, en aangeboren glaucoom zijn andere mogelijk geassocieerde oogziekten. Ook ondiepe oogkassen kunnen problemen met het gezichtsvermogen veroorzaken. Vaak heeft de patiënt door deze ooggerelateerde problemen een visuele handicap en kan hij blind of (ernstig) slechtziend zijn.
Vingers en tenen
Patiënten met het Apert-syndroom hebben zwemvliezen of samengesmolten (gefuseerde) vingers en tenen. De ernst van de fusie varieert. Normaal gesproken zijn minstens drie vingers aan elke hand en drie tenen aan elke voet met elkaar versmolten. In de meest ernstige gevallen zijn alle vingers en tenen gefuseerd. Ook kan een abnormale kromming aan de vinger of teen ontstaan, gekend als "clinodactylie". Minder vaak hebben patiënten met deze aandoening extra vingers of tenen (polydactylie).
Bijkomende symptomen
Bijkomende symptomen van het Apert-syndroom omvatten gehoorverlies, ongewoon veel zweten (hyperhidrosis), een kort gestalte, slaapapneu (slaapstoornis met tijdelijk gestopte ademhaling), hartafwijkingen, spijsverteringsproblemen, urinewegproblemen, een vette huid met ernstige acne, stukken wenkbrauw die ontbreken, samengesmolten nekwervels (cervicale wervels), en recidiverende oorinfecties of sinusinfecties die gepaard kunnen gaan met een gespleten gehemelte (schisis).
Diagnose en onderzoeken
Lichamelijk onderzoekDe diagnose van het syndroom gebeurt door middel van een grondig lichamelijk onderzoek op basis van de zichtbare symptomen. De arts merkt zo de craniosynostose op.
Diagnostisch onderzoek
Een röntgenonderzoek (radiografisch onderzoek), een gehooronderzoek, en een CT-scan en MRI-scan zijn andere nuttige onderzoeksinstrumenten. De bevestiging van de diagnose gebeurt aan de hand van een genetisch onderzoek.
Differentiële diagnose
De differentiële diagnoses omvatten:
- het Carpenter-syndroom (kleeblattschadel, klaverbladschedelmisvorming)
- het syndroom van Crouzon (craniofaciale dysostose)
- het Jackson-Weiss-syndroom: Afwijkingen aan hoofd, gezicht en voeten
- het Pfeiffer-syndroom (voortijdige fusie van schedelbotten)
- het Beare-Stevenson-syndroom
- het Saethre-Chotzen-syndroom (afwijkingen aan hoofd en gezicht, handen en voeten en hersenen)
Behandeling
Dit syndroom is niet te genezen. Een multidisciplinaire behandeling is echter wel mogelijk waarbij een kinderarts, een orthodontist, een neuroloog, een KNO-arts, een oogarts, en andere zorgverstrekkers samenwerken. Craniofaciale chirurgie kan worden toegepast om de schedel- en gezichtsafwijkingen te verhelpen. Ook correctieve chirurgie voor de gefuseerde vingers en tenen is aanbevolen. Oogdruppels en/of oogzalf kunnen de symptomen van droge ogen voorkomen of verlichten. Hierbij dient de patiënt de oogdruppelrichtlijnen strikt te volgen. Een CPAP-apparaat helpt bij slaapproblemen. Antibiotica worden voorgeschreven voor sinus- en oorinfecties. Het chirurgisch plaatsen van oorbuisjes kan bij sommige kinderen noodzakelijk zijn. Tot slot kan een tracheostomie noodzakelijk zijn bij kinderen met ernstig obstructief slaapapneu.Prognose aandoening
De schedeloperatie dient idealiter op jonge leeftijd te worden uitgevoerd, omdat de kans op een normale intellectuele ontwikkeling afneemt naarmate het kind ouder wordt. Zelfs met de operatie kunnen sommige hersenstructuren zich mogelijk slecht ontwikkelen. Het is vaak gunstiger voor kinderen om in de thuisomgeving te blijven in plaats van in een instelling, omdat dit hun intellectuele capaciteiten kan bevorderen. Sommige kinderen met het Apert-syndroom presteren zelfs uitstekend op de universiteit. Andere patiënten kunnen echter ernstiger getroffen zijn, wat betekent dat de levensverwachting kan variëren tussen de patiënten.Complicaties
- Dubbelzien: Bij afwijkingen in de oogspieren of zenuwen kan dubbelzien optreden. Dit kan ontstaan door een onjuiste werking van de spieren die normaal gesproken de ogen in dezelfde richting laten bewegen.
- Nystagmus: Ongecontroleerde, snelle bewegingen van de ogen kunnen voorkomen bij problemen met de oogspieren of zenuwen die verantwoordelijk zijn voor de oogbewegingen.
- Scheelzien: Problemen met de zenuwen die de oogspieren aansturen kunnen leiden tot scheelzien, waarbij de ogen niet goed op hetzelfde punt gericht zijn.
- Oogspierverlamming: Afwijkingen in de zenuwen of spieren kunnen leiden tot gedeeltelijke of volledige verlamming van een of meer oogspieren, wat de oogbewegingen ernstig kan beïnvloeden.
- Visuele beperking: Bij ernstige gevallen kunnen de oogspierproblemen leiden tot een verminderd gezichtsvermogen of een wazig beeld, vooral als de ogen niet goed kunnen samenwerken.
Preventie
Momenteel zijn er geen effectieve preventiemethoden voor het Apert-syndroom, omdat het meestal het gevolg is van spontane genmutaties. Vroegtijdige diagnose en een multidisciplinaire benadering kunnen helpen om de symptomen te beheersen en de levenskwaliteit van de patiënten te verbeteren. Genetisch advies kan nuttig zijn voor ouders met een familiegeschiedenis van craniosynostose of verwante aandoeningen.Praktische tips voor het leven met Apert-syndroom
Apert-syndroom is een zeldzame genetische aandoening die wordt gekarakteriseerd door de vroege sluiting van de schedelnaden, wat leidt tot een abnormale schedelvorm en gezichtsafwijkingen. Het heeft vaak ook invloed op de handen en voeten, die vaak samengegroeide vingers en tenen vertonen. Het is belangrijk om goed te weten hoe je het beste omgaat met de uitdagingen die dit syndroom met zich meebrengt, zowel fysiek als emotioneel.Volg een gezonde levensstijl
Gezonde eet- en leefgewoonten zijn belangrijk voor iedereen, maar ze kunnen nog belangrijker zijn wanneer je leeft met een aandoening zoals Apert-syndroom. Een evenwichtig voedingspatroon ondersteunt niet alleen je algehele gezondheid, maar kan ook bijdragen aan een betere wondgenezing en het behouden van energie voor fysieke activiteiten. Het vermijden van een alcohol en het beperken van suiker kan helpen om ontstekingen en andere complicaties te verminderen.Zorg voor je fysieke gezondheid en mobiliteit
Bij Apert-syndroom kan er sprake zijn van bewegingsbeperkingen door de samengegroeide handen en voeten. Fysiotherapie is vaak een belangrijk onderdeel van de behandeling om de mobiliteit te verbeteren en de functie van de handen en voeten te ondersteunen. Het kan nuttig zijn om samen te werken met een fysiotherapeut die kan helpen bij het ontwikkelen van een bewegingsplan dat je helpt om zo veel mogelijk van je dagelijks leven zelfstandig uit te voeren.Neem contact op met specialisten voor chirurgische ingrepen
Chirurgie kan nodig zijn om de fysieke afwijkingen die het gevolg zijn van Apert-syndroom te corrigeren, zoals het openen van de schedelnaden en het verbeteren van de vinger- en teenfunctie. Het is belangrijk om regelmatig medische controles te hebben en samen te werken met chirurgen, zoals een plastisch chirurg of een orthopedisch specialist, die ervaring hebben met het behandelen van dit syndroom. Ze kunnen ook begeleiding bieden over de beste tijdstippen voor chirurgische ingrepen en postoperatieve zorg.Let op je mentale gezondheid
Leven met Apert-syndroom kan een emotionele uitdaging zijn, vooral als het gaat om zelfbeeld en sociale interacties. Het is belangrijk om aandacht te besteden aan je mentale gezondheid en je emoties te uiten. Het kan nuttig zijn om met een psycholoog of therapeut te praten over de gevoelens die je hebt over je uiterlijk en de impact die de aandoening heeft op je leven. Ondersteuning van familie, vrienden en lotgenoten kan je helpen om met de sociale en emotionele aspecten van het syndroom om te gaan.Volg een zorgplan voor regelmatige medische controleafspraken
Aangezien Apert-syndroom veel invloed kan hebben op verschillende systemen in het lichaam, is het van cruciaal belang om regelmatig medische controles te hebben. Dit kan onder meer het monitoren van de hersenen, het controleren van de gezichtsontwikkeling, en het evalueren van de voortgang van de hand- en voetfunctie omvatten. Het ontwikkelen van een zorgplan met je artsen en het naleven van controleafspraken is essentieel om complicaties vroegtijdig op te sporen en een optimale gezondheid te behouden.Misvattingen rond Apert-syndroom
Apert-syndroom is een zeldzame genetische aandoening die invloed heeft op de schedel-, hand- en voetontwikkeling. De aandoening wordt veroorzaakt door een mutatie in het FGFR2-gen en leidt tot vroegtijdige vergroeiing van de schedelbeenderen en vaak ook samenvoeging van vingers en tenen. Ondanks de medische kennis die beschikbaar is, bestaan er nog steeds veel misvattingen over deze aandoening.Apert-syndroom is altijd erfelijk
Hoewel Apert-syndroom door een genetische mutatie wordt veroorzaakt, betekent dit niet dat het altijd erfelijk is. In de meeste gevallen ontstaat de mutatie spontaan tijdens de vroege embryonale ontwikkeling, zonder dat een van de ouders de mutatie draagt. Erfelijke overdracht is wel mogelijk als een ouder met Apert-syndroom de genetische mutatie doorgeeft aan een kind, maar dit is minder vaak het geval.Alle patiënten met Apert-syndroom hebben eenzelfde uiterlijk
Hoewel bepaalde kenmerken zoals een verkorte schedel, uitpuilende ogen en vergroeiingen aan de handen en voeten veel voorkomen, kunnen de uiterlijke kenmerken sterk variëren tussen patiënten. De ernst van de botvergroeiingen en bijkomende kenmerken zoals kaakafwijkingen of huidproblemen kunnen per individu verschillen.Apert-syndroom beïnvloedt enkel de botstructuur
Naast de opvallende schedel- en ledemaatafwijkingen kan Apert-syndroom ook invloed hebben op andere delen van het lichaam. Patiënten kunnen gehoorproblemen, ademhalingsmoeilijkheden en darmproblemen ervaren. Daarnaast kunnen cognitieve en neurologische uitdagingen optreden, hoewel deze niet bij iedereen even ernstig zijn.Alle patiënten met Apert-syndroom hebben een verstandelijke beperking
Hoewel sommige patiënten met Apert-syndroom een milde tot matige verstandelijke beperking kunnen hebben, geldt dit niet voor iedereen. De mentale ontwikkeling hangt af van meerdere factoren, waaronder de ernst van de schedelvergroeiing en de mate van vroege medische interventies. Met tijdige chirurgie en passende begeleiding kunnen veel patiënten een normale intelligentie en goede cognitieve vaardigheden behouden.Er is geen behandeling mogelijk voor Apert-syndroom
Hoewel Apert-syndroom niet genezen kan worden, bestaan er diverse chirurgische en therapeutische interventies om de kwaliteit van leven te verbeteren. Chirurgie kan de schedelvergroeiingen corrigeren om hersengroei en ademhaling te ondersteunen. Daarnaast kunnen hand- en voetoperaties helpen om functionaliteit te verbeteren. Orthodontische en logopedische begeleiding kan verdere ondersteuning bieden bij spraak- en voedingsproblemen.Patiënten met Apert-syndroom kunnen geen zelfstandig leven leiden
Met de juiste medische zorg en ondersteuning kunnen veel patiënten met Apert-syndroom een zelfstandig leven leiden. De mate van zelfredzaamheid hangt af van de ernst van de aandoening en de beschikbare behandelingen. Sommige patiënten kunnen volledig onafhankelijk functioneren, terwijl anderen baat hebben bij extra ondersteuning in het dagelijks leven. Vroege interventie en multidisciplinaire zorg kunnen een aanzienlijk verschil maken in de levenskwaliteit van patiënten met Apert-syndroom.© 2016 - 2025 Miske, het auteursrecht van dit artikel ligt bij de infoteur. Zonder toestemming is vermenigvuldiging verboden. Deze informatie is van informatieve aard en geen vervanging voor professioneel medisch advies. Per 2021 gaat InfoNu verder als archief, artikelen worden nog maar beperkt geactualiseerd.
 Jackson-Weiss-syndroom: Afwijkingen aan gezicht en voetenHet Jackson-Weiss-syndroom is een zeldzame aandoening gekenmerkt door opvallende afwijkingen van het hoofd en gezicht do…
Jackson-Weiss-syndroom: Afwijkingen aan gezicht en voetenHet Jackson-Weiss-syndroom is een zeldzame aandoening gekenmerkt door opvallende afwijkingen van het hoofd en gezicht do…
 Syndactylie: met elkaar vergroeide tenen of vingersVergroeide tenen, zwemvliezen tussen uw vingers, het komt vaker voor dan u misschien denkt en het heet syndactylie. Hoe…
Syndactylie: met elkaar vergroeide tenen of vingersVergroeide tenen, zwemvliezen tussen uw vingers, het komt vaker voor dan u misschien denkt en het heet syndactylie. Hoe…
 Syndroom van Crouzon: Voortijdige fusie van schedelbottenHet syndroom van Crouzon is een genetische aandoening die zich kenmerkt door de voortijdige fusie van bepaalde schedelbo…
Syndroom van Crouzon: Voortijdige fusie van schedelbottenHet syndroom van Crouzon is een genetische aandoening die zich kenmerkt door de voortijdige fusie van bepaalde schedelbo…
 Muenke-syndroom: Afwijkingen aan hoofd (schedel) en gezichtBij het Muenke-syndroom, een erfelijke aandoening, wordt een baby geboren met vroegtijdig gesloten kroonnaden in de sche…
Muenke-syndroom: Afwijkingen aan hoofd (schedel) en gezichtBij het Muenke-syndroom, een erfelijke aandoening, wordt een baby geboren met vroegtijdig gesloten kroonnaden in de sche…
 Nager-syndroom: Craniofaciale afwijkingHet Nager-syndroom is een uiterst zeldzame erfelijke aandoening gekenmerkt door craniofaciale misvormingen in combinatie…
Nager-syndroom: Craniofaciale afwijkingHet Nager-syndroom is een uiterst zeldzame erfelijke aandoening gekenmerkt door craniofaciale misvormingen in combinatie…
 Hoe ontstaat een zonnesteek en hoe kom ik er vanaf?Een zonnesteek wordt meestal pas ontdekt wanneer het te laat is. Dit gebeurt vaak wanneer iemand te lang in de zon heeft…
Hoe ontstaat een zonnesteek en hoe kom ik er vanaf?Een zonnesteek wordt meestal pas ontdekt wanneer het te laat is. Dit gebeurt vaak wanneer iemand te lang in de zon heeft…
Gerelateerde artikelen
Bronnen en referenties
- http://emedicine.medscape.com/article/941723-clinical#showall
- http://emedicine.medscape.com/article/941723-differential
- http://emedicine.medscape.com/article/941723-overview#showall
- http://emedicine.medscape.com/article/941723-workup#showall
- http://ghr.nlm.nih.gov/condition/apert-syndrome
- http://rarediseases.org/rare-diseases/apert-syndrome/
- http://www.apert.org/apert.htm
- http://www.erfelijkheid.nl/ziektes/apert-syndroom
- http://www.webmd.com/children/apert-syndrome-symptoms-treatments-prognosis?page=3
- http://www.webmd.com/children/apert-syndrome-symptoms-treatments-prognosis?page=2
- http://www.webmd.com/children/apert-syndrome-symptoms-treatments-prognosis
- https://www.nlm.nih.gov/medlineplus/ency/article/001581.htm
- Afbeelding bron 1: Marco Mayer, Wikimedia Commons (CC BY-SA-4.0)
Miske (4.039 artikelen)
Laatste update: 26-02-2025
Rubriek: Mens en Gezondheid
Subrubriek: Aandoeningen
Bronnen en referenties: 13
Laatste update: 26-02-2025
Rubriek: Mens en Gezondheid
Subrubriek: Aandoeningen
Bronnen en referenties: 13
Per 2021 gaat InfoNu verder als archief. Het grote aanbod van artikelen blijft beschikbaar maar er worden geen nieuwe artikelen meer gepubliceerd en nog maar beperkt geactualiseerd, daardoor kunnen artikelen op bepaalde punten verouderd zijn. Reacties plaatsen bij artikelen is niet meer mogelijk.
Medische informatie…
Deze informatie is van informatieve aard en geen vervanging voor professioneel medisch advies. Raadpleeg bij medische problemen en/of vragen altijd een arts.
Deze informatie is van informatieve aard en geen vervanging voor professioneel medisch advies. Raadpleeg bij medische problemen en/of vragen altijd een arts.